

Freiburg und London, 31. Januar 2023
Mitschrift der Live-Debatte:
Am 26. Januar 2023 diskutierten deutsche und britische Experten über politische Optionen zur Beendigung der Verschwendung von Forschungsergebnissen in klinischen Studien in Deutschland, wobei sie die nationale #MakeItPublic-Strategie Großbritanniens als Bezugspunkt nahmen.
Die Online-Veranstaltung wurde von TranspariMED organisiert, von Cochrane Deutschland moderiert und von Consilium Scientific ausgerichtet. Nachfolgend finden Sie das deutschsprachige Transkript, das aus Gründen der Klarheit und Kürze leicht bearbeitet wurde. Übersetzt mit www.DeepL.com/Translator (kostenlose Version).
Sie können auch ein Video der Veranstaltung auf YouTube ansehen (auf englisch).

TRANSKRIPT
Till Bruckner, TranspariMED [00:00:01]
Hallo zusammen. Vielen Dank, dass Sie heute bei uns sind. Mein Name ist Till Bruckner, von der TranspariMED-Kampagne von Consilium Scientific. Wir werden darüber sprechen, ob und wie das britische System zur Transparenz klinischer Studien in anderen Ländern umgesetzt werden kann.
Diese Diskussion ist wirklich für ganz Europa relevant, denn in Europa haben wir derzeit drei verschiedene Regulierungssysteme für klinische Studien, die einen großen Einfluss auf die Transparenz haben.
- Erstens, und das ist das Beste, haben wir das System für Arzneimittelprüfungen [CTIMPs], bei dem die Veröffentlichung der Ergebnisse gesetzlich vorgeschrieben ist. Und es gibt auch eine wirksame Durchsetzung durch die Regulierungsbehörden. Ich denke, dass sich vor allem die deutschen Aufsichtsbehörden dadurch ausgezeichnet haben, dass sie dies ernst genommen haben.
- Der zweite Punkt betrifft die Versuche mit Medizinprodukten, bei denen es ebenfalls eine gesetzliche Verpflichtung gibt, die Ergebnisse [einiger Versuche mit Medizinprodukten] zu veröffentlichen. Aber ich denke, wenn wir uns anschauen, wie die Durchsetzung und Einhaltung der Vorschriften aussehen wird, dann wird das eine wirklich große Herausforderung sein.
- Und dann haben wir noch alle anderen Studien. Es gibt also Studien, die weder Arzneimittelprüfungen noch die speziellen Medizinprodukteprüfungen sind, die unter die Medizinprodukteverordnung fallen, und zwar in Deutschland und in allen anderen großen EU- Ländern. Zurzeit gibt es keinerlei gesetzliche Verpflichtung, die Ergebnisse dieser Studien zu veröffentlichen. Und natürlich gibt es auch keine Durchsetzung, weil es keine Vorschriften gibt, die durchgesetzt werden können.

Sources: https://Iink.springer.com/articIe/10.1007/s00103−022−03631 −x
https://www.transparimed.org/singIe−post/cIinicaI−triaI−reguIation−europe
Wenn wir uns also die Arzneimittelprüfungen ansehen, können wir in den kommenden Jahren mit einer sehr strengen Einhaltung der Vorschriften rechnen. Bei den Prüfungen von Medizinprodukten und allen "anderen" Prüfungen wird es schwierig werden. Wir haben heute Daniel Strech auf dem
Podium, der die IntoValue2-Studie geleitet hat, aus der hervorgeht, dass etwa 30 % der von akademischen Einrichtungen in Deutschland durchgeführten klinischen Prüfungen ihre Ergebnisse nie veröffentlichen werden. Es gibt also eine enorme Forschungsverschwendung in diesem Bereich.
Und in diesem Zusammenhang halte ich das britische Modell für eine wirklich interessante potenzielle Lösung für dieses Problem, die auch in anderen europäischen Ländern übernommen werden könnte. Wie funktioniert es? Auf der linken Seite sehen Sie den Ablauf einer normalen klinischen Prüfung: Die Prüfung wird geplant, dann wird sie zur Ethikgenehmigung eingereicht, sie wird registriert, dann wird die Prüfung durchgeführt und am Ende werden die Ergebnisse veröffentlicht. Das britische Modell folgt diesem normalem Prozess.
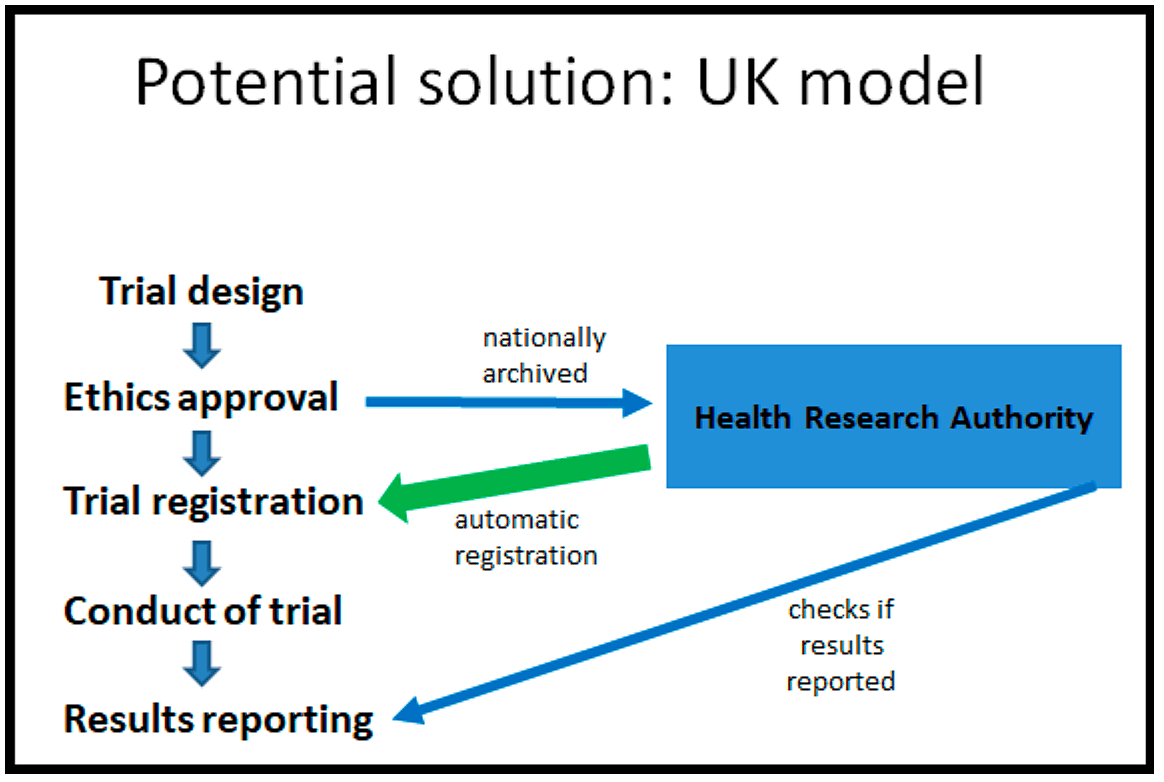
Aber das neue System, das jetzt eingeführt wird, sieht vor, dass nach der Ethikgenehmigung die Dokumentation der Ethikgenehmigung von der Gesundheitsforschungsbehörde landesweit archiviert wird. Jemand in der Gesundheitsforschungsbehörde registriert dann die klinische Prüfung für Sie auf der Grundlage der ethischen Unterlagen.
Dann führen Sie die Prüfung wie gewohnt durch. Sie machen die Ergebnisse öffentlich. Und danach führt die Gesundheitsforschungsbehörde eine doppelte Kontrolle durch. Haben Sie die Ergebnisse veröffentlicht? Wenn Sie die Ergebnisse nicht veröffentlicht haben, wird man Sie kontaktieren und Sie daran erinnern, die Ergebnisse zu veröffentlichen.
Es ist also ein sehr einfaches System, das einige große Vorteile hat.
- Der erste ist natürlich, wie Sie unten sehen können, die Veröffentlichung von Informationen. Es deckt alle klinischen Prüfungen ab. Es überwindet diese künstlichen regulatorischen Unterscheidungen, denn jede klinische Prüfung muss ethisch genehmigt werden, und es stellt sicher, dass 100 % dieser Prüfungen registriert werden und 100 % von ihnen auch ihre Ergebnisse veröffentlichen. Es handelt sich also um ein wirklich wasserdichtes System, das alle Prüfungen abdeckt.
- Der zweite Vorteil besteht darin, dass Transparenz zur Norm wird. Es gibt also sehr, sehr klare Regeln, die für alle klinischen Prüfungen gelten. Die Studienteilnehmer wissen also ganz genau, was von ihnen erwartet wird. Außerdem gibt es sehr effiziente Verfahren, die die Teilnehmer automatisch zur Transparenz führen.
- Der dritte Vorteil besteht darin, dass Transparenz leicht gemacht wird, und das bedeutet weniger Bürokratie für medizinische Forscher, weniger Papierkram für medizinische Forscher, damit sich die Wissenschaftler auf das konzentrieren können, was sie am besten können, nämlich die Wissenschaft.
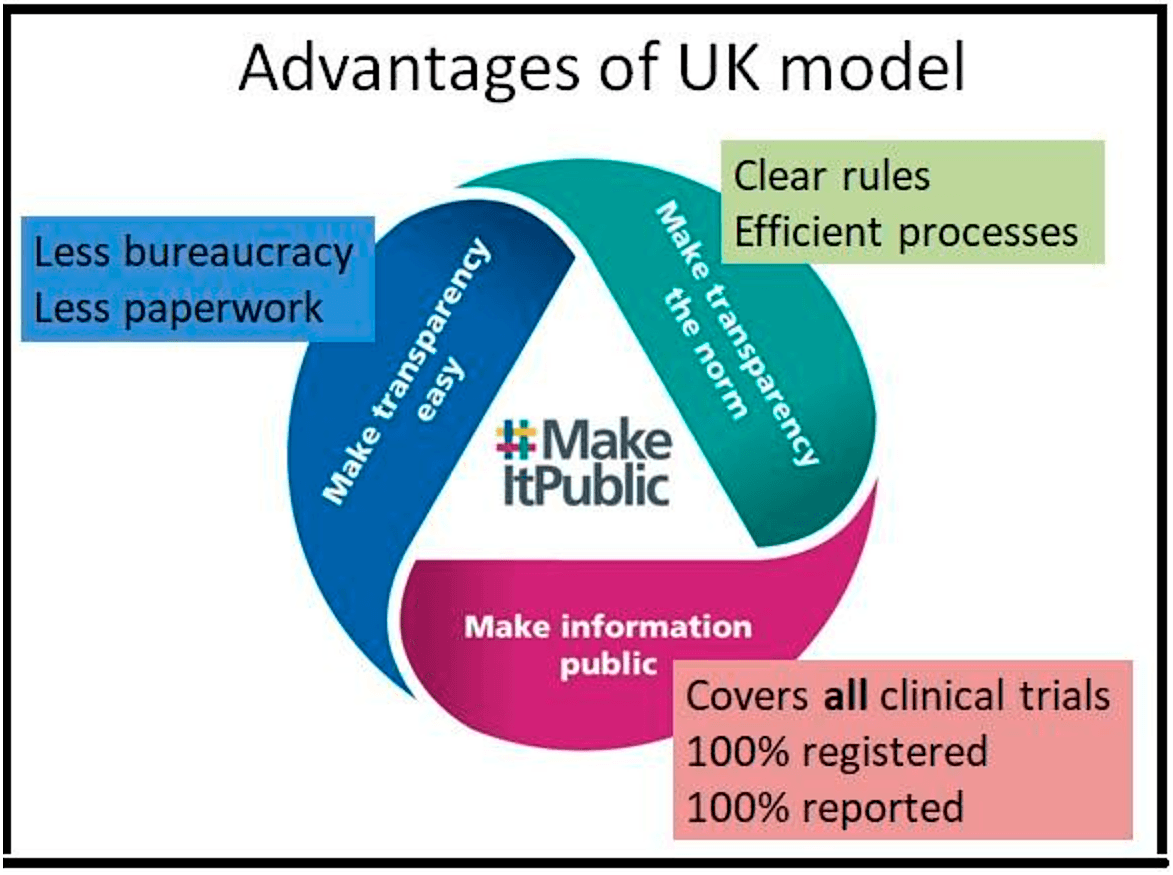
Kann dies auch anderswo geschehen? Und das ist das Thema der heutigen Sitzung. Wir haben eine großartige Expertenrunde zusammengestellt.
- Zunächst werden wir erfahren, wie es dem Vereinigten Königreich gelungen ist, dieses System so gut zu verwirklichen. Martin Smith, der dem parlamentarischen Ausschuss angehörte, der diesen Plan entwickelt hat, wird über die politischen Beweggründe für dieses System sprechen. Dann haben wir Naho Yamasaki, die bei der Health Research Research Authority arbeitet; sie war federführend bei der Entwicklung des Systems. Und schließlich haben wir C. Mark Taylor, den ehemaligen Vorsitzenden des ISRCTN-Studienregisters, der über die praktischen Probleme bei der Umsetzung dieses Systems sprechen wird.
- Zweitens werden wir Kommentare aus Deutschland hören. Wir haben Matthias Perleth vom HTA Verein, Daniel Strech vom QUEST Center for Reproducible Research, Stefan Sauerland vom Deutschen EBM Netzwerk und Christoph Stein, Vorstandsmitglied von Transparency International Deutschland. Sie werden jeweils ganz kurz die Möglichkeiten und Hindernisse für die Nutzung dieses Systems in Deutschland erörtern.
- Danach haben wir eine Stunde Zeit für eine freie Diskussion, die von Valerie Labonte von Cochrane Deutschland moderiert wird.
Martin Smith, ehemaliger Sachbearbeiter im britischen Parlament
Im Jahr 2018 unterstützte ich als Mitarbeiter den Ausschuss für Wissenschaft und Technologie des britischen Unterhauses im Parlament. Das ist ein formeller Ausschuss des britischen Parlaments. Es handelt sich dabei um eine parteiübergreifende Gruppe von Abgeordneten, die vom Parlament ernannt werden, um die Arbeit der britischen Regierung in Bezug auf wissenschaftliche und technologische Fragen zu überprüfen. Ich denke, es gibt eine Entsprechung im Ausschusssystem des Deutschen Bundestages, aber im Vereinigten Königreich konzentrieren sich die Sonderausschüsse nicht auf die Überprüfung von Rechtsvorschriften. Der Schwerpunkt liegt ausschließlich auf der Kontrolle der Regierung, und sie erstellen Berichte, zu denen die Regierung schriftlich Stellung nehmen muss.
Wir würden sagen, dass sie eher sehr einflussreich als mächtig sind. Sie haben zwar keine Exekutivgewalt, aber sie verfügen über großen politischen Einfluss. Im Jahr 2018 führte der Ausschuss eine Untersuchung zur Integrität der Forschung im weitesten Sinne durch. Im Anschluss daran veröffentlichte er einen eigenen Bericht speziell zur Transparenz klinischer Studien. Ich war für beide Arbeiten zuständig, d. h. ich habe die schriftlichen Eingaben analysiert, mit den Experten in diesen Bereichen Kontakt aufgenommen und den Bericht für den Ausschuss verfasst. Seitdem bin ich weitergezogen. Ich arbeite jetzt für den Wellcome Trust in London, daher spreche ich hier in meiner persönlichen Eigenschaft und berufe mich auf meine eigenen Überlegungen aus dieser Zeit.
Aber wenn Sie gestatten, möchte ich mit einem Zitat aus dem Bericht des Ausschusses von 2018 beginnen: "Die Transparenz klinischer Studien ist ebenso sehr eine Frage des politischen Willens wie eine technische Frage". Und ich denke, das fasst den Weg des Vereinigten Königreichs hier zusammen und ist ein guter Rahmen für die Kommentare, die ich machen möchte.
Der erste Punkt, den ich ansprechen möchte, ist, dass es eigentlich die Aktivisten waren, die die Gelegenheit für eine Überprüfung in diesem Bereich geschaffen haben. Sie waren es, die den Ausschuss direkt dazu veranlassten, sich speziell mit diesem Thema zu befassen. Die ursprüngliche Untersuchung der Kommission bezog sich auf die Integrität der Forschung, auf Dinge wie Betrug und Irrtum, Publikationsverzerrungen und dergleichen mehr. Es gab tatsächlich eine kleine Anzahl von Eingaben zu dieser Untersuchung, unter anderem von Till und AllTrials und einigen anderen, die den Standpunkt vertraten, dass die Transparenz klinischer Studien ein viel größeres Problem darstellt als all diese Dinge, oder zumindest viel häufiger auftretende Probleme hat als ein Thema wie Betrug, auch wenn diese sehr bekannt und problematisch sein mögen.
Aber als ich ursprünglich die Untersuchung zur Integrität der Forschung vorbereitete, hatte ich die Transparenz nicht als einen besonderen Bereich vorgesehen. Als Reaktion auf diese Eingaben sahen Aktivisten wie Till die Möglichkeit, diesem Thema politischen Nachdruck zu verleihen, und so kam es zur Arbeit des Ausschusses.
Ich sollte auch sagen, dass dies nicht das erste Mal war, dass sich der Ausschuss mit der Transparenz von Studien befasst hat. Sein Vorgänger hatte sich fünf Jahre zuvor, im Jahr 2015 [Anm.: eigentlich war es 2013], mit einer anderen Gruppe von Abgeordneten befasst, natürlich mit diesem politischen Wechsel. Sie hatten sich ebenfalls mit diesem Thema befasst und einige sehr vernichtende Schlussfolgerungen gezogen, die jedoch nicht wirklich umgesetzt worden waren. Auch dies war ein zusätzlicher Vorteil für den Ausschuss, wenn er sich erneut mit diesem Thema befasst. Die Möglichkeit, frühere Arbeiten weiterzuverfolgen, insbesondere wenn etwas ignoriert wurde, ist für einen Ausschuss sehr reizvoll.
Einer der Unterschiede zwischen 2013 und 2018 war die Einrichtung der Gesundheitsforschungsbehörde (Health Research Authority, HRA) im Jahr 2014, und Sie werden später von der Gesundheitsforschungsbehörde hören. Was dem Ausschuss jedoch auffiel, war, dass es zu den Aufgaben der HRA gehört, die Integrität der Forschung zu fördern. Was der Ausschuss jedoch aus den Eingaben und den öffentlichen Anhörungen mitbekam, war, dass die HRA keine Strategie zu haben schien, um dies zu tun. Wenn überhaupt, dann schien sie eher einen passiven Ansatz zur
Förderung der Transparenz zu verfolgen, als Verbesserungen voranzutreiben, und sie maß ihre Leistung nicht an den Veränderungen, die sie bewirkte.
Ich denke, das ist bezeichnend, weil die Existenz des HRA einen offensichtlichen Eigentümer für dieses Problem geschaffen hat, bis skizziert wurde, wie sie jetzt in das System passen, das sehr gut funktioniert. Ein weiteres Puzzleteil war die Tatsache, dass es eine enorme Zunahme von Regeln und Vorschriften gab, die eine Registrierung und Berichterstattung vorschreiben. Till hat das im deutschen Kontext angesprochen. Aber im Vereinigten Königreich wurden diese nicht durchgesetzt.
Der gesetzliche und regulatorische Rahmen war also bereits vorhanden. Jetzt verbinden sich diese beiden Dinge auf interessante Weise. In diesem Fall gibt es einige Dinge, die die Aufmerksamkeit der Abgeordneten auf sich ziehen, wie ich aus meiner langjährigen Zusammenarbeit mit ihnen weiß. Das eine ist die Möglichkeit, etwas, das im Grunde genommen klar und deutlich zu erklären ist, mit Nachdruck zu fordern. Das zweite ist die Möglichkeit, dass sie das tun und dadurch tatsächlich eine Veränderung bewirken. Beides ist attraktiv: auf der richtigen Seite der Geschichte zu stehen, aber auch in der Lage zu sein, tatsächlich etwas zu bewirken. Unabhängig davon, wie zynisch man zu Politikern steht, ist die überwiegende Mehrheit der Politiker in diesem Geschäft tätig, um Veränderungen zu bewirken.
Bei der Transparenz klinischer Studien schien es sich also um ein lösbares Problem zu handeln, ein klares: Der gesetzliche Rahmen ist jetzt besser, aber es fehlte der politische Wille, ihn wirklich zu überwinden.
Ich denke, es sollte auch erwähnt werden, dass der Ausschuss damals feststellen konnte, dass einige der Antworten der HRA wie ziemlich schwache Ausreden wirkten. Die Position schien nicht sehr vertretbar zu sein, wenn man darauf hinwies, dass es kein Budget für diese Dinge gab. Versuche, zu ermitteln, um welche Art von Geld es sich handelt, deuteten darauf hin, dass es im Großen und Ganzen gar nicht so teuer war, insbesondere angesichts des Umfangs und der Bedeutung dieses Themas. Ich denke, dass insbesondere der Ausschussvorsitzende den Spielraum riechen konnte, sich wirklich damit zu befassen und regelmäßig Folgemaßnahmen zu ergreifen, wenn die HRA nicht daran interessiert war, den Empfehlungen des Ausschusses nachzukommen, indem sie einfach sagte, dass dies überhaupt nicht nötig sei.
Und tatsächlich endet mein Teil der Geschichte mit dem Bericht des Ausschusses, mit seinen Empfehlungen und der Antwort der Regierung. Die wohl wichtigste Empfehlung des Ausschusses war, dass die Regierung die Gesundheitsforschungsbehörde auffordern sollte, eine neue Strategie zu entwickeln, um die Verbesserung der klinischen Versuche und der Transparenz wirklich voranzutreiben, und sie gegebenenfalls mit den entsprechenden Haushaltsbefugnissen auszustatten, um in diesem Bereich wirklich etwas zu bewirken.
Ich freue mich, sagen zu können, dass die Regierung dieses Angebot sehr wohl angenommen hat, und die Antwort der HRA darauf war genau die richtige. Ich schließe meine Ausführungen mit einem Zitat des damaligen HRA-Vorsitzenden, das lautete: "Bei der Förderung der Forschung war Transparenz immer ein wichtiger Bestandteil unserer Arbeit. Der Ausschuss hat uns vor die Herausforderung gestellt, nicht mehr nur bewährte Verfahren zu fördern, sondern Verbesserungen voranzutreiben. Wir nehmen diese Herausforderung an." Das ist Musik für einen Ausschuss.
Das war definitiv das Richtige, und seit 2018 ist viel passiert, weil die HRA sich dieser Herausforderung gestellt hat. Ich denke, der Ausschuss war bereit, noch viel weiter zu gehen, wenn es nötig gewesen wäre, aber das war nicht nötig. Also herzlichen Glückwunsch an die Aktivisten, die dieses Thema überhaupt erst auf die Tagesordnung des Ausschusses gebracht haben, und dann an die HRA, die so gut reagiert hat.
Naho Yamasaki, Britische Behörde für Gesundheitsforschung (HRA) [00:14:08]
Ich werde die Geschichte dort fortsetzen, wo Martin aufgehört hat. Bevor ich den Faden von Martins Vortrag weiterverfolge, möchte ich ein wenig Hintergrundwissen über die Health Research Authority vermitteln. Für diejenigen unter Ihnen, die uns nicht kennen: Wir wurden vor 11 Jahren gegründet, um die Forschung im Gesundheits- und Sozialwesen in ganz Großbritannien zu rationalisieren und zu koordinieren. Wir sind eine unabhängige Einrichtung des Ministeriums für Gesundheit und Soziales, so dass die Regierung ihre gesamte Verantwortung in diesem Bereich der Regulierung an uns abgetreten hat. Die meisten unserer Aufgaben betrafen die in England durchgeführte Forschung, aber wir arbeiteten eng mit Schottland, Wales und Nordirland zusammen, um ein britisches System und die Schlüsselkomponenten für unsere Arbeit zu schaffen.
Was das Genehmigungsverfahren für die Forschung betrifft, so bieten wir einen UK-weit koordinierten Dienst für die Überprüfung von Forschungsethik. Wir unterstützen die 60 Forschungsethikausschüsse. Im Auftrag des Nationalen Gesundheitsdienstes prüfen wir auch, ob die vorgeschlagene Studie in England und Wales mit den rechtlichen und verwaltungstechnischen Anforderungen übereinstimmt. Und wir unterstützen auch einen Ausschuss, der Studien überprüft, die identifizierbare Patientendaten ohne Zustimmung verwenden wollen. Dies sind also die Aufgaben, die wir im Rahmen unserer operativen Unterstützung wahrnehmen.
Und wie Martin bereits erwähnt hat, sind wir auch gesetzlich verpflichtet, die Transparenz in der Forschung zu fördern. Nach dem Bericht des Ausschusses und der Aufforderung, eine Strategie zu entwickeln, haben wir uns an die Arbeit gemacht und dabei festgestellt, dass die damals verfügbaren Daten zeigen, dass die Transparenz im Vereinigten Königreich ziemlich schlecht ist.
Nur etwa 70 % der klinischen Prüfungen wurden in einem anerkannten Register registriert, obwohl dies eine Bedingung für die Genehmigung durch die Forschungsethikkommission war. Außerdem meldeten 75 % der klinischen Arzneimittelstudien ihre Ergebnisse nicht rechtzeitig an das [EudraCT]- Register, wobei die Quote bei anderen Studien noch niedriger war. Und trotz dieses Vorschlags in den [Ethik-]Anträgen hat die Mehrheit der klinischen Prüfungen die Teilnehmer nicht über die Ergebnisse informiert.
Um dies zu ändern, haben wir eine Strategie entwickelt. Zunächst haben wir eine beratende Gruppe eingesetzt, die einen Entwurf für eine Strategie erarbeitet hat, die sich mit den vier Säulen der Transparenz befasst. Das sind die Registrierung, die Veröffentlichung der Ergebnisse, die Rückmeldung der Ergebnisse an die Teilnehmer sowie die gemeinsame Nutzung von Forschungsdaten und Gewebe. Wir begannen mit der Beratung zu dieser Strategie. Dies ist eine wichtige Komponente bei dem Versuch, die Zustimmung des Sektors und der Gemeinschaft zu erhalten, denn wir wollten eine ehrgeizige Strategie entwickeln, die uns zu der Vision einer vollständigen Transparenz im gesamten System führt, aber mit Mitteln, die sinnvoll und handhabbar sind und die die Zustimmung des gesamten Sektors finden.
Daher haben wir von Juni bis September eine dreimonatige öffentliche Konsultation durchgeführt und gleichzeitig eine Online-Umfrage sowie eine Reihe öffentlicher Workshops veranstaltet. Wir haben auch eine Reihe von Webinaren für die Mitglieder unserer Forschungsethikkommission durchgeführt und eine Reihe von Fokusgruppen mit Patienten abgehalten, was meiner Meinung nach ebenfalls sehr wichtig ist. Wir haben eine ganze Reihe von Workshops mit unseren Mitarbeitern durchgeführt, weil wir erkannt haben, dass wir nicht nur die Unterstützung des Sektors brauchen, sondern auch das Personal, um den Wandel zu ermöglichen.
Über die Online-Umfrage haben wir etwas mehr als 700 Antworten von Einzelpersonen und Organisationen auf die Fragen erhalten, die wir in Bezug auf den Strategieentwurf gestellt hatten. Wir haben all diese Antworten ausgewertet und die Daten für die Strategie #MakeItPublic zusammengestellt, die im Juli 2020 veröffentlicht wurde.
Die Vision, die wir formuliert haben, ist ganz einfach. Sie besteht darin, dass vertrauenswürdige Informationen über die Gesundheits- und Sozialfürsorgeforschung zum Nutzen aller öffentlich zugänglich gemacht werden. Um diese Vision zu verwirklichen, sind wir im Rahmen der Strategie zehn
Verpflichtungen eingegangen, die sich auf drei übergreifende Ziele stützen: Wir werden Transparenz einfach machen, Transparenz zur Norm machen und Informationen öffentlich machen. Ich denke, dies wurde durch die Erkenntnis untermauert, dass die Forschergemeinschaft durch den Dialog, den wir geführt haben, und den Konsultationsprozess den Wunsch geäußert hat, offen und transparent über ihre Forschung zu sein. Im Großen und Ganzen gibt es also eine große Unterstützung für Transparenz. Wir sind uns aber auch bewusst, dass es viele Dinge gibt, die die Transparenz erschweren, und deshalb wollten wir mit der Strategie eine Kombination von Ansätzen bieten, einschließlich eines Systems von Prozessänderungen, der Bereitstellung von Lernhilfen und der Hervorhebung guter und schlechter Praktiken, und mit diesen verschiedenen Ansätzen wollten wir erreichen, dass die Erfüllung der Anforderungen an die Forschungstransparenz einfach zu dem gehört, was wir alle tun.
Ich möchte nur auf ein paar Dinge eingehen, die wir während der Konsultation getan haben, um es einfach und zur Norm zu machen.
- Die Forscher sagten uns, dass die Anforderungen an die Transparenz nicht immer klar sind, ist das also der einfache Weg? Zu einem großen Teil. Eines der ersten Dinge, die wir getan haben, war also, die Anforderungen zu klären. Wir haben unsere Leitlinien für unsere Websites, Antragsformulare und Genehmigungsschreiben überprüft und aktualisiert. So sind unsere Erwartungen an die Forscher und Sponsoren viel klarer geworden.
- Wir haben auch einen Fahrplan entwickelt. Er zeigt den gesamten Ablauf einer klinischen Prüfung mit den wichtigsten Punkten, an denen Antragsteller die Anforderungen an die Forschungstransparenz erfüllen müssen, damit sie im Voraus planen können. Dabei handelt es sich um einen Prototyp eines Fahrplans, den wir weiterentwickeln wollen.
- Wir haben auch ein Lernmodul erstellt, das erklärt, wie man eine Zusammenfassung der Forschungsergebnisse in einfacher Sprache verfasst, und wir denken über weitere Arten von Leitlinien in diesem Bereich nach.
Das sind also die Änderungen, die wir unterstützen wollen, damit Forscher leichter verstehen, was sie tun müssen, wann sie es tun müssen und wie sie Transparenz gut umsetzen können.
Um die Transparenz in der Strategie zur Norm zu machen, haben wir gesagt, dass wir gute Praktiken belohnen und würdigen und auch schlechte Leistungen hervorheben werden, dass wir mit anderen Akteuren im System, wichtigen Interessengruppen, Geldgebern, anderen Regulierungsbehörden und Verlegern zusammenarbeiten werden, um sicherzustellen, dass unsere Erwartungen an die Forschungstransparenz konsistent sind und übereinstimmen. Und man kann mit Fug und Recht behaupten, dass wir in diesem Bereich noch viel mehr tun müssen. Aber das ist unser Bestreben - und auch, dass wir Maßnahmen ergreifen werden, wenn jemand seine Transparenzanforderungen nicht erfüllt. Es geht also um eine Kombination aus Anstößen und wahrscheinlich etwas stärkeren Maßnahmen, um Veränderungen herbeizuführen.
Um Erfolge zu feiern und bewährte Praktiken hervorzuheben, haben wir eine Konferenz abgehalten und im November 2021 unseren ersten Jahresbericht veröffentlicht, der als Aufzeichnung zur Verfügung steht, falls Interesse besteht. Wir haben auch eine Kampagnengruppe gegründet, die wichtige Interessenvertreter aus dem gesamten System zusammenbringt, um Veränderungen anzustoßen. Diese Interessenvertreter haben einige dieser Konferenzen und den Bericht maßgeblich mitgestaltet und die Gestaltung von drei Workshops unterstützt, die wir im März dieses Jahres durchführen wollen.
Die Idee ist, dass wir die Arbeit mit der Kampagnengruppe und der breiteren Stakeholder- Gemeinschaft fortsetzen, um unsere Arbeit an der Angleichung der Erwartungen fortzusetzen und die Bedeutung der Forschungstransparenz weiter zu verdeutlichen.
Der andere Bereich, mit dem wir uns befassen, ist, wie ich bereits sagte, das Ergreifen von Maßnahmen und das Nachdenken darüber, wie wir als Regulierungsbehörde am besten damit umgehen können, wenn Erwartungen nicht erfüllt werden. Und damit wir überhaupt etwas unternehmen können, müssen wir uns auf korrekte Leistungsdaten verlassen können. Und im Moment haben wir keine vollständigen Informationen, auf deren Grundlage wir handeln können.
Und wir haben Systeme eingerichtet, die uns das ermöglichen. So. Im September 2021 haben wir eine neue Art der Einreichung von Studienabschlussberichten eingeführt. Es war schon immer vorgeschrieben, dass alle Studien 12 Monate nach dem Ende der Studie einen Abschlussbericht bei einer Forschungsethikkommission einreichen. Bis dahin gab es jedoch weder ein Standardformular noch eine genaue Vorgabe, was in diesem Bericht enthalten sein muss. Um dies zu ändern, haben wir einen Standarddatensatz erstellt, den die Teilnehmer uns in diesem Abschlussbericht vorlegen müssen, um uns darüber zu informieren, wie sie die Anforderungen an die Forschungstransparenz erfüllt haben oder ob und wie sie sie erfüllt haben. Das betrifft die Registrierung, die Veröffentlichung der Ergebnisse, die Versorgung der Teilnehmer und die Weitergabe von Forschungsdaten und Gewebe.
Es gibt also vier Säulen der Transparenz, über die ich gesprochen habe. Wir haben auch verlangt, dass eine Zusammenfassung der Ergebnisse als Teil des Abschlussberichts eingereicht wird, und wir haben damit begonnen, diese auf unserer Website zu veröffentlichen, um die Zusammenfassung der Studienpläne zu ergänzen, die wir als Teil des Antragsverfahrens erhalten.
Außerdem sind wir eine Partnerschaft mit einem britischen Register namens ISRCTN eingegangen, um die Registrierung klinischer Prüfungen im Namen von Sponsoren zu besprechen. Wir beginnen mit klinischen Arzneimittelstudien [CTIMPs] und planen, dies in Zukunft weiter auszubauen. Die Idee ist, die Sponsoren zu entlasten, weil wir die Daten an das Register liefern. Das ist aber auch ein weiterer Weg, um sicherzustellen, dass es in Zukunft eine 100 %ige Abdeckung gibt.
Das ist also die Arbeit, die wir geleistet haben und weiter leisten. Und ich denke, man kann mit Fug und Recht behaupten, dass es noch mehr Arbeit zu tun gibt. Aber es gibt definitiv den Willen und das Engagement der Organisation, diese Praxis im gesamten System zu verwirklichen.
C. Mark Taylor, Ex-Vorsitzender des ISRCTN-Studienregisters [00:25:05]
In den letzten zehn Jahren war ich Vorsitzender des Unternehmens, dem das ISRCTN-Register [klinische Studien] gehört. Das ist ein etwas ungewöhnliches Arrangement. Schon früh wurde beschlossen, dass das Register nicht Teil einer öffentlichen Einrichtung sein sollte und dass es am besten wäre, es unabhängig zu betreiben, und zwar unter dem Eigentum eines Unternehmens, das über einen Vertrag läuft.
Bevor ich nach meiner Pensionierung den Vorsitz dieses Unternehmens übernahm, war ich Beamter im britischen Ministerium für Gesundheit und Soziales und arbeitete zehn Jahre lang für den Chief Medical Officer und den Direktor für Forschung und Entwicklung am Aufbau der Strukturen, zu denen auch die Health Research Authority gehört. Ich erinnere mich, dass ich die Grundsatzdokumente verfasst habe, die zur Gründung der Behörde führten.
Die speziellen Themen, auf die ich jetzt eingehen möchte, knüpfen an das an, was Martin und Naho gesagt haben. Ich möchte damit niemanden in Deutschland entmutigen, aber viele der von Martin beschriebenen Elemente reichen weit zurück, und zwar bis in die Anfänge meiner Zeit im Gesundheitsministerium. Es gab also eine Zusammenarbeit zwischen dem System der Forschungsethikkommissionen und dem Genehmigungsdienst der MHRA [britische Arzneimittelbehörde], die der Health Research Authority vorausging. Und es bestand ein starkes Interesse daran, jemandem die Verantwortung für die Förderung der Transparenz zu übertragen.
Das führte zu dem Gesetz, mit dem 2014 die Health Research Authority (HRA) eingerichtet wurde, die die Aufgabe hat, die Transparenz im öffentlichen Interesse zu fördern. Ich glaube, die HRA ist die einzige öffentliche Einrichtung im Vereinigten Königreich, die eine solche Pflicht hat, trotz all der anderen Forschungsbereiche. Und als die Transparenzstrategie 2010 veröffentlicht wurde, geschah dies parallel zur Erprobung eines kombinierten verbesserten Genehmigungsverfahrens, das die Aktivitäten des Ethikkommissionssystems mit der Regulierung von Arzneimitteln und Produkten verknüpfte. Und wie Naho bereits sagte, wurde der Aufgabenbereich nicht nur auf zugelassene Arzneimittel und Geräte, sondern auf alle Arten von Forschung ausgeweitet, die im öffentlichen
Interesse stattfinden und eine Ethikkommission passieren können. Das war also der Hintergrund für die Konzentration auf klinische Versuche.
Und es waren nicht nur die Health Research Authority und die MHRA. Ich meine, seit Anfang der 2000er Jahre haben sich unsere Chief Medical Officers in der Weltgesundheitsversammlung für die Förderung der Transparenz eingesetzt, zusammen mit der Registrierung von Studien und der Berichterstattung. Der Grund für diesen Vorstoß war die Notwendigkeit, die wissenschaftliche Reaktion auf Pandemien und Bedrohungen der globalen öffentlichen Gesundheit zu koordinieren. Es geht hier also sowohl um ein internationales Anliegen, ich würde sagen, um die Solidarität mit anderen Menschen, die an einer transparenten Wissenschaft in der ganzen Welt interessiert sind, als auch um die Verantwortung der Gesundheitsforschungsbehörde in England, auf die, wie ich meine, berechtigte Kritik unserer Parlamentarier zu reagieren.
Als das ISRCTN-Register im Jahr 2000 seine Arbeit aufnahm, begann es als internationales Register mit starker Unterstützung der britischen Regierung durch den Chief Medical Officer und in der Erwartung, dass es mit anderen im WHO-System zusammenarbeiten würde.
Das ist es also. Sie haben mich gebeten, über die Umsetzung zu sprechen, aber ich will damit sagen, dass die Umsetzung der Strategie, die wir jetzt erleben, lange vorher begonnen hat. Die Bedeutung der Strategie liegt darin, dass sie die Dinge wirklich gut zusammenführt. Und wie Martin und Naho sagten, hat die HRA den Auftrag, in diesem Bereich eine Führungsrolle zu übernehmen, und sie hat definitiv eine Führungsrolle außerhalb des Bereichs der Regulierung übernommen, denn neben den internationalen Unternehmen, aktiven und lizenzierten Produkten gibt es im Vereinigten Königreich einen großen Sektor mit Wohltätigkeitsorganisationen, die die Gesundheitswissenschaften fördern. Sie kennen vielleicht unsere Association of Medical Research Charities, 150 Mitglieder mit über einer Milliarde pro Jahr, die vor nicht allzu langer Zeit eine offene Forschungsplattform eingerichtet hat, um den Austausch von Ergebnissen zu fördern. Als Nahos Vorbereitungen für das Charta der Strategie im Gange waren, gab es bereits eine Reihe von Wohltätigkeitsorganisationen, die 2017 bei der Weltgesundheitsorganisation eine gemeinsame Erklärung über die Angleichung der Richtlinien zur Veröffentlichung von [Studien-]Ergebnissen unterzeichnet hatten.
Und so war das Transparenzforum, das die HRA eingerichtet hatte, ein sehr aufgeschlossener Ort, an dem das ISRCTN-Register einige der Daten hinter den Fehlern, die Martins im Parlament hervorgehoben sah, veröffentlichen konnte. ISRCTN hat als Hintergrund der Strategie die Daten über die Situation nicht nur im Vereinigten Königreich, sondern auch in anderen forschungsaktiven Ländern veröffentlicht. Und ich muss sagen, dass wir uns sehr aktiv mit der Leistung der deutschen Universitäten und Aufsichtsbehörden verglichen haben, und tatsächlich ist die Leistung der französischen Universitäten miserabel. Besonders besorgniserregend fand ich, dass insbesondere die französischen Universitäten schlecht dafür sorgten, dass die Ergebnisse ihrer klinischen Versuche an die Öffentlichkeit gelangten.
Es ging also um das Vertrauen der Öffentlichkeit und darum, den Forschern die Arbeit zu erleichtern, und um die Arbeit des Registers in Verbindung mit den Prüfverfahren. [Das System war lange Zeit sehr kundenorientiert, und man stellte ein stabiles Expertenteam ein, das die Leute bei ihrer recht schwierigen Aufgabe, Studien zu registrieren, beriet. Ich sage "schwierig", weil zu diesem Zeitpunkt nicht viele Sponsoren daran interessiert waren. Wir boten also ausführliche Beratung an und stellten dem Register Funktionen zur Verfügung, die die Registrierung, Aktualisierung und Berichterstattung ermöglichten, und schickten auch Mahnungen an Personen, die die Fristen nicht eingehalten hatten. Das waren wirklich wichtige Aufgaben für ein Register, nicht nur die Pflege der Daten.
Und auf internationaler Ebene muss noch viel mehr getan werden, um die Datensätze und Datenflüsse anzugleichen. Ich freue mich, dass das HRA und auch das National Institute for Health Research im Vereinigten Königreich sich für einen durchgängigen Datenfluss interessieren, der es den Menschen leicht macht, die Beschreibung ihrer Arbeit in die verschiedenen betroffenen Systeme hinein- und wieder herauszuschicken.
Ich meine, die Registrierung und Berichterstattung sollte ein Nebeneffekt von ordnungsgemäß geführten Datenverwaltungs- und Managementsystemen bei den Sponsoren sein, und diese sollten mit den Regulierungsbehörden abgestimmt werden, und davon sind wir noch weit entfernt.
Sie haben mich also gebeten, ein paar Erfahrungen zu sammeln, und ich werde mich nicht mehr lange damit aufhalten, aber es gibt hier eine Menge zu bedenken.
Zunächst einmal habe ich betont, dass es lange gedauert hat, die Kultur zu ändern, und wir sind noch nicht so weit. Aber die große Veränderung kommt, wenn die Sponsoren auf Transparenz bestehen und interne Systeme einrichten und Forschungsmanager einstellen, die ihre Forschung dabei unterstützen, mit der Herausforderung umzugehen, sich während der gesamten Dauer einer Forschungsaktivität daran zu erinnern, was sie in Bezug auf Transparenz tun sollten.
Und der zweite Punkt ist die Unterstützung. Es dauert lange, ein Netzwerk von Forschungsmanagern aufzubauen. Und das Research Integrity Office im Vereinigten Königreich [UKRIO] fördert seit über zehn Jahren neben dem HRA die Debatte über Transparenz. Und Martin hat vorhin schon auf die Hauptstoßrichtung der Untersuchung hingewiesen, die von allen Parlamentariern aufgegriffen wurde. Aber viele Institutionen im Vereinigten Königreich bieten ihrem Forschungsmanagement und ihren Forschungsteams immer noch sehr schwache, inkonsistente Unterstützung. Sie unterstützen keine Leute, die in der Lage sind, von einem Ende einer Aktivität zum anderen zu wissen, was ihnen fehlt, und sie aktualisieren und berichten Elemente.
Und der letzte Punkt sind die Forschungsdatensysteme, denn sie haben noch nicht wirklich mit der Politik in der Praxis Schritt gehalten. Und das ist ein Fehler, den wir meiner Meinung nach gemacht haben. Ich glaube, wir haben einen Fehler gemacht, als wir die Partnerschaft zwischen dem HRA und dem ISRCTN eingeführt haben. Bevor wir das in die Wege geleitet haben, hätten wir viel länger mit Datenmanagern in der Industrie und an den Universitäten sowie mit Geldgebern über die Mechanismen sprechen sollen, wie man Daten problemlos von einem System in ein anderes überführen kann, und wie man die Prozesse und Definitionen angleichen kann, was bedeuten würde, dass man keine neue Diskussion über den Registrierungsdatensatz führen muss.
Ich möchte nur sagen, dass ich als Forscher Eigentümer dieses Datensatzes bin. Es ist eine korrekte Beschreibung dessen, worüber ich zu berichten gedenke, wenn die Forschung abgeschlossen ist. Der Haken an der Sache ist, und das ist mein letzter Punkt, dass es im Vereinigten Königreich immer noch viele Forscher gibt, die darauf bestehen, sich weiterhin bei ClinicalTrials.gov zu registrieren, weil sie das in Frankreich gewohnt sind. Jeder registriert sich bei ClinicalTrials.gov, wenn er das tut. Ich bin ein großer Bewunderer der US-amerikanischen National Library of Medicine, aber es ist wirklich nicht die Aufgabe der NLM, einen Registrierungs- und Meldedienst für die gesamte entwickelte Welt zu betreiben, auch nicht für die Menschen hier, die durchaus in der Lage sind, diese Aufgabe selbst ordnungsgemäß zu erledigen.
Und wir wissen aus Erfahrung, dass, wenn man sich bei der falschen Registrierstelle anmeldet und keine Folgemaßnahmen ergreift, dies der beste Weg ist, um eine unzureichende Berichterstattung oder eine schlechte Qualität der Berichterstattung, ein geringes Engagement der Sponsoren und wirklich schlechte Beziehungen zu den Menschen, die an der Studie teilgenommen haben, sicherzustellen. Das bedeutet also, dass die Forschung aus Sicht der Teilnehmer und der Gesellschaft im Allgemeinen umsonst ist. Das Vertrauen der Menschen, die ein echtes Interesse an den Ergebnissen unserer klinischen Forschung haben, ist stark erschüttert.
Valerie Labonte, Cochrane (Moderatorin) [00:38:23]
Valerie Labonte hier von Cochrane Deutschland. Und jetzt werden wir von den deutschen Experten hören, wie ein System zur Transparenz klinischer Studien in Deutschland umgesetzt werden könnte. Die Fragen lauten also: Welche Anpassungen müssten vorgenommen werden, um das System an den deutschen Kontext anzupassen, und was wird wahrscheinlich die größte Herausforderung sein und wie könnte man diese bewältigen? Ich möchte Matthias Perleth bitten, zuerst zu antworten.
Matthias Perleth, HTA Verein [00:38:58]
Hallo zusammen und vielen Dank, dass Sie dieses Treffen einberufen haben. Ich bin der Vorsitzende der Gesellschaft für Health Technology Assessment. Das ist nur ein sehr kleiner Verein, so dass wir wahrscheinlich nicht in der Lage sind, den gesamten Prozess in Deutschland zu organisieren. Aber wir können natürlich alles tun, was wir können, um die Aktivitäten in Deutschland irgendwie zu unterstützen.
Es ist nicht leicht, auf diese beiden Fragen zu antworten. Lassen Sie mich nur einige kurze Bemerkungen dazu machen. Wie wir am Beispiel der Autos sehen können, die die deutschen Straßen blockieren, kennen wir in Deutschland jedes Detail über diese Autos. Eine Registrierung und Transparenz ist also offensichtlich möglich. Im Gesundheitswesen gibt es jedoch weniger Transparenz, und das hat wahrscheinlich ganz andere Gründe.
Ein gutes Beispiel dafür, dass Transparenz möglich ist und eine Registrierungspflicht Realität werden könnte, ist ein Trauma- und Implantatregister. Etwa im Jahr 2025 wird es für Hüft- und Knieimplantate in Kraft treten. Sie müssen ab 2025 verpflichtend registriert werden. Und diese Entwicklung ist insofern bedeutsam, als dass die Registerpflicht in Deutschland Realität werden könnte. Aber natürlich erfordert dies, wie gerade gesagt wurde, eine Art Entwicklungs- und Kontrolländerung, da dieses Register bereits vor etwa zehn Jahren eingerichtet wurde, aber nur auf freiwilliger Basis.
Aber das deutsche Ministerium hat diese Entwicklung auch als Reaktion auf die Situation bei Medizinprodukten und Europa im Allgemeinen und auf Skandale mit fehlerhaften Geräten aufgegriffen, um die Sicherheitslage in Deutschland zu verbessern. Das war also der auslösende Faktor für die Einrichtung eines obligatorischen Registers. Wir könnten versuchen, eine Kampagne in Deutschland zu unterstützen. Ich denke, was wir brauchen, ist ein nationales Bündnis.
Und ich bin sehr froh, dass zum Beispiel auch ein Vertreter des Deutschen Netzwerks für Evidenzbasierte Medizin [DnEBM] hier anwesend ist. Wir bräuchten wahrscheinlich Ethikkommissionen, da sie auch im britischen System eine entscheidende Rolle spielen. Wir bräuchten Leute aus dem deutschen Parlament, Abgeordnete, aus dem Gesundheitsministerium, aus der Forschung, von Geldgebern und Sponsoren und von medizinischen Universitäten. Ich denke, viele der Studien, die nie veröffentlicht werden, sind wahrscheinlich von Prüfärzten initiierte Studien.
Erschwerend kommt hinzu, dass wir keine deutsche Behörde für Gesundheitsforschung oder ein Äquivalent dazu haben. Wir müssen also andere Wege finden, um diese nationale Allianz zusammenzustellen und unsere Politiker irgendwie darauf aufmerksam zu machen. Wie Martin eingangs sagte, wird es eine Frage des politischen Willens sein. Und wir als kleinere Gesellschaft können versuchen, diesen Prozess zu unterstützen.
Daniel Strech, QUEST [00:43:23]
Es wären sicherlich eine Menge technischer Anpassungen notwendig, wenn wir in Deutschland so etwas wie den Prozess kopieren wollten, den Sie im Begriff sind zu installieren und der in Großbritannien bereits teilweise umgesetzt wurde. Und ich werde mich nicht zu all diesen eher technischen Aspekten äußern, sondern aus einer übergeordneten Perspektive, den eher konzeptionellen und politischen Anpassungen.
Ich würde sagen, dass sich alle europäischen Länder im Verfahren für Arzneimittelprüfungen [CTIMPs] annähern, da alle Genehmigungen für Arzneimittelprüfungen von den Ethikkommissionen an die Behörden gehen, die verpflichtet sind, die klinische Prüfung im europäischen Register für klinische Prüfungen registrieren zu lassen. Wir haben also diesen Registrierungsprozess in gewisser Weise bereits etabliert, aber nur für Arzneimittelprüfungen. Und das sind nur etwa ein Drittel aller interventionellen Studien.
Die politische Anpassung müsste darin bestehen, dass die deutschen Forschungsethikkommissionen, die nicht nur Arzneimittelstudien, sondern auch alle anderen interventionellen Studien genehmigen, über ein Verfahren verfügen müssen, mit dem sie eine zentrale Stelle über die genehmigten Studien informieren können. Und das haben wir noch nicht. Wie Matthias bereits erwähnte, haben wir keine HRA, also müssten wir sehen, wie diese zentrale Stelle aussehen könnte.
Es gibt mehrere Gremien, die offensichtlich eine Rolle spielen würden, aber wir haben kein zentrales Gremium, das die Führung übernimmt, die, wie Sie erwähnten, jetzt wichtig ist. Wir haben diesen Dachverband für alle deutschen Ethikkommissionen [AKEK].
Und außerdem, ich meine, das war nur der Teil der Registrierung, um alle Studien zu registrieren. Zweitens die Berichterstattung über die Ergebnisse, was bei Arzneimittelstudien oder gemäß den gesetzlichen Bestimmungen relativ einfach ist. Aber für alle nichtmedikamentösen Studien bräuchten wir wirklich eine Änderung der Politik in Deutschland, um dies zu gewährleisten.
Was die wichtigsten Herausforderungen angeht, so würde ich sagen, dass eine Herausforderung darin bestehen könnte, dass wir nicht alle vorhandenen Ressourcen nutzen, die wir haben. Ich denke, in vielerlei Hinsicht müssen wir uns nicht neu erfinden, nicht nur, weil es bereits ein echtes Beispiel aus dem Vereinigten Königreich gibt, sondern auch aufgrund der Ressourcen, die wir in Deutschland haben.
Wir haben die verschiedenen Parteien und Gremien, die ich gerade erwähnt habe, die im Prinzip in der Lage wären, all die verschiedenen Versuche zu überwachen, und sie könnten im Prinzip einige Verfahren zur Weiterleitung von Informationen einführen, diese zentralisieren und öffentlich machen. Aber neben den Organisationen ist es meiner Meinung nach auch sehr wichtig, alle Software und Codes zu nutzen, die es uns ermöglichen, Informationen aus den Registern und über die Berichterstattung über die Ergebnisse auf eine automatisierte Weise zu erhalten. Es ist also nicht notwendig, dass die Leute wirklich manuell nach Registrierungsergebnissen suchen. Es gibt bereits eine Menge Software, die es uns ermöglicht, Informationen aus dem deutschen Register für klinische Studien [DRKS], aus ClinicalTrials.gov und aus den europäischen Registern für klinische Studien [EudraCT & CTIS] zu erhalten. Und es ist wichtig, dass die Organisationen in Deutschland diese bereits existierende Software nutzen, um alles so einfach wie möglich zu machen, da Sie sie als einen wichtigen Teil Ihrer Initiative hervorgehoben haben.
Nicht zuletzt besteht die Herausforderung darin, die relevanten Entscheidungsträger an einen Tisch zu bekommen. Also ich würde den Leuten, die heute hier zusammensitzen, von Cochrane oder dem EBM-Netzwerk sagen, der gemeinsame Bundesausschuss, um den es geht [G-BA], ist auch ein bisschen vertreten. Es sind verschiedene Parteien. Auch mein [QUEST] Institute for Responsible Research. Ich denke, dass es Leute gibt, die gerne eine Allianz bilden würden, um vielleicht auch ein bisschen Druck zu machen.
Aber wir brauchen auch die Bündnisse der Entscheidungsträger, um die Führung zu übernehmen. Und ich hoffe, dass dies heute eine wichtige Veranstaltung ist, um dies ein wenig in Gang zu bringen. Ich danke Ihnen.
Stefan Sauerland, Deutsches EBM-Netzwerk (DnEBM) [00:49:03]
Danke, dass Sie dieses Treffen organisiert haben. Die deutsche Einrichtung, die ich leite, die Abteilung für Nichtarzneimittel [des IQWIG], hat mit all diesen Problemen zu kämpfen. Wir hatten zum Beispiel einen Bericht, bei dem es schwierig war, zu einer Schlussfolgerung zu kommen, weil wir so viele registrierte Studien hatten, die keine Ergebnisse gebracht hatten. Aber ich bin auch für das deutsche Netzwerk für evidenzbasierte Medizin hier. Und da ich viele Jahre an der Universität gearbeitet habe, kenne ich auch die andere Seite, denn ich habe viele klinische Studien durchgeführt und etwa ein Dutzend chirurgische Studien registriert, von denen einige gut und einige weniger gut dokumentiert sind.
Ich stimme dem zu, was bisher gesagt worden ist. Das Hauptproblem ist, dass es diese verschiedenen Arten von Studien gibt, wie Till sie bereits kategorisiert hat.
Wir haben die Arzneimittelstudien, und diese werden nicht nur den Ethikkommissionen in Deutschland gemeldet, sondern müssen auch vom Bundesinstitut für Arzneimittel und Medizinprodukte [BfArM] genehmigt werden, das auch das Deutsche Register für klinische Prüfungen [DRKS] führt. Sie wissen von Prüfungen, die unter die [europäische] Medizinprodukteverordnung oder das deutsche Pendant fallen.
Das Hauptproblem sind jedoch andere Studien, die nicht unter die Arzneimittel- oder Medizinprodukteverordnung fallen, denn diese werden nur von der Ethik-Kommission eingesehen, und sie können die Prüfer nur schlecht davon überzeugen, die Ergebnisse zu melden.
Daher denke ich, dass das BfArM hier die Hauptrolle bei der Ersetzung der britischen Gesundheitsforschungsbehörde spielen würde, denn es hat bereits all diese Studien zu Arzneimitteln und viele der Studien zu Geräten. Und wenn wir die lokalen Ethikkommissionen davon überzeugen könnten, dass sie alle ihre Studien, die in die dritte Kategorie fallen, an das BfArM schicken, könnte das BfArM ein Verzeichnis aller Studien führen. Und daraus würde dann so etwas wie eine Überwachungsliste für alle deutschen Geräte entstehen. So wüssten sie von allen Studien, die zumindest ein randomisiertes Design haben. Wir können auch über nicht-randomisierte Studien sprechen, die registriert worden sind. Aber für den Anfang würde ich nur mit den randomisierten kontrollierten Studien beginnen.
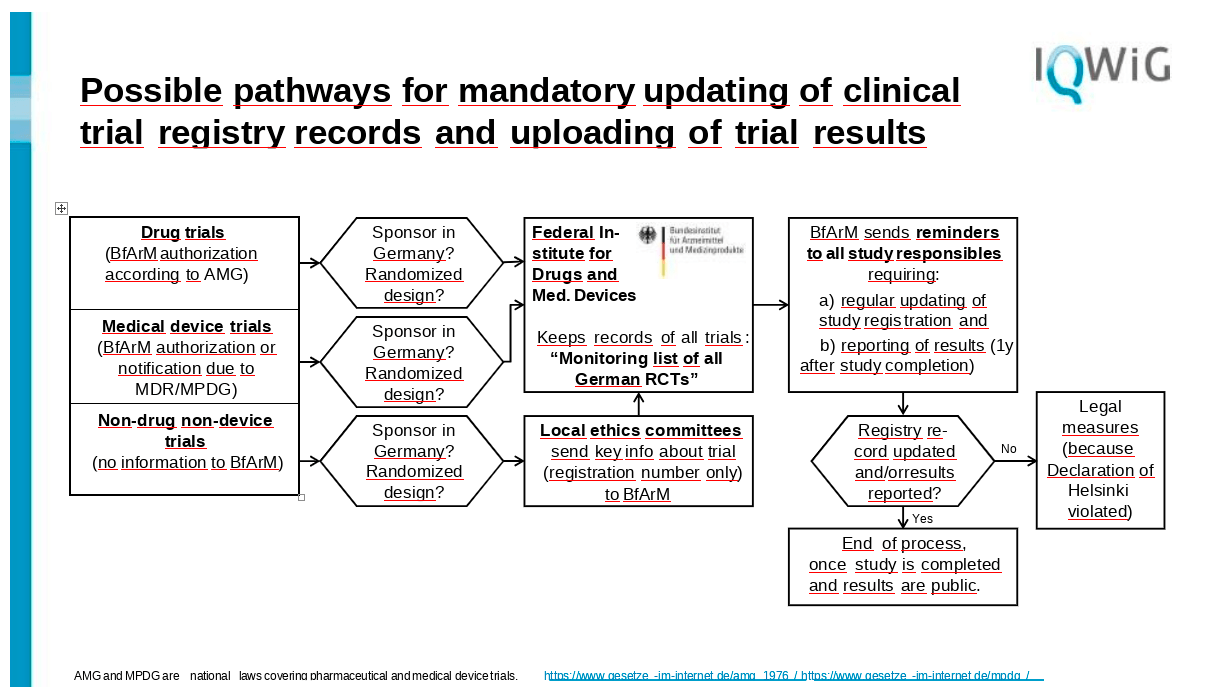
Und das ist natürlich nur notwendig, wenn der Sponsor in Deutschland ist, denn die anderen Studien, wenn nur ein deutsches Zentrum an der Studie teilnimmt, muss die ganze Angelegenheit in dem Land geregelt werden, in dem der Hauptsponsor sitzt, und jetzt kommt der schwere Teil.
Das BfArM müsste alle Verantwortlichen anmahnen, und sie müssten mindestens einmal im Jahr die Studienregistrierung aktualisieren. Und natürlich, wenn die Studie das Ende des Rekrutierungszeitraums vor einem Jahr erreicht hat, wäre eine Berichterstattung über die Ergebnisse in irgendeiner Form erforderlich.
Dafür ist das BfArM meiner Meinung nach ganz gut geeignet, weil es auch diese maßgebliche Rolle in der deutschen Forschung hat und das würde nicht unbemerkt bleiben, um es mal ganz offen zu sagen. Also die Forscher können dann natürlich entweder dem Aktualisierungswunsch des BfArM folgen, und am Ende werden die Ergebnisse gemeldet.
Aber wenn das nicht der Fall ist, und das ist der kritischste Punkt, dann müsste man erst einmal rechtliche Mittel in der Hand haben, um die Ermittler unter Druck zu setzen. Natürlich könnte man sich an die Chefs der Prüfer wenden, z. B. an den Dekan der Universität, oder an die Ethikkommission, die der Studie einmal zugestimmt hat, damit der Prüfer keine neue Studie beginnen kann, bevor er die vorherige Studie ordnungsgemäß abgeschlossen hat.
Und das schwerwiegendste Element wäre, dass sie sich an die Ärztekammer wenden könnten, um zu verlangen, dass die Ärzte, die die Studie durchführen, die Deklaration von Helsinki befolgen. In gewisser Weise ist das alles schon im Gesetz verankert, denn zumindest in Deutschland gibt es in den Satzungen der Ärztekammern für meinen Bereich die gesetzliche Vorgabe, dass wir uns alle an die Deklaration von Helsinki halten müssen. Und das bedeutet, dass wir die Ergebnisse von klinischen Studien veröffentlichen müssen.

Es gibt also eine mögliche Rechtsgrundlage, die ein Argument für die Kontaktaufnahme mit den Prüfärzten sein könnte. Das Hauptproblem wäre also, dass wir einige Ressourcen brauchen, vielleicht bei den Ethikkommissionen, aber hauptsächlich beim BfArM. Die technischen Fragen, die Daniel gerade angesprochen hat, die automatische Aufzeichnung oder die automatische Verbindung zwischen den Aufzeichnungen und den Veröffentlichungen und so weiter, würden keine gesetzliche Änderung erfordern. Es würde nur voraussetzen, dass irgendein Beamter im Gesundheitsministerium, na ja, eine Richtlinie erlässt, damit das BfArM diese Rolle im deutschen Gesundheitswesen für die Aktualisierung der Studienregister und die Bereitstellung der Studienergebnisse übernimmt. Das war's.
Christoph Stein, Vorstandsmitglied von Transparency International Deutschland [00:56:19]
Da alle meine Vorredner bereits über die aktuelle Situation gesprochen haben und ich denke, dass sie sehr gut beschrieben wurde, habe ich nur ein paar Fragen. Zunächst einmal bin ich mir nicht ganz sicher, warum ich hier sitze. Warum ist Deutschland heute das Ziel dieses Panels und nicht die EU? Warum sitzt Ursula von der Leyen nicht hier? Ich denke, meine Erfahrung, und ich spreche mehr oder weniger als klinischer Forscher, ist, dass wir normalerweise versuchen, die Gesetze einzuhalten, und wir sind sehr empfänglich für Gesetze in Deutschland und wir sind meistens empfänglich für die EU-
Gesetze. Ich denke also, dass wir meiner Meinung nach nicht nur auf Deutschland abzielen sollten, sondern auf die gesamte EU.
Aber dann habe ich noch ein paar Fragen an meine Kollegen aus dem Vereinigten Königreich. Zunächst einmal bin ich mir nicht ganz sicher, was sie mit Veröffentlichung meinen. Wir haben in den letzten Jahren im Zusammenhang mit dieser Pandemie viel über Veröffentlichungen gesprochen, und wir haben gesehen, dass es ein großes Problem mit Veröffentlichungen gibt. Wir müssen darüber reden, ob die Leute nur Preprints oder von Fachleuten geprüfte Veröffentlichungen lesen. Ich denke also, dass dies für uns klar definiert werden muss.
Und noch eine sehr detaillierte Frage an meine Kollegen aus dem Vereinigten Königreich: Wie sind Ihre Erfahrungen mit der Zahl der Studien? Es liegt auf der Hand, dass die Registrierung und all diese Anforderungen eine Belastung für die Studiendurchführenden darstellen, auch für die Sponsoren. Und meine Frage lautet: Haben Sie jemals einen Rückgang der Zahl der Studien aufgrund dieser zusätzlichen Belastung festgestellt?
Das ist wirklich alles, was ich zu diesem Zeitpunkt zu sagen habe.
Valerie Labonte, Cochrane Deutschland (Moderatorin) [00:59:05]
Vielen Dank für Ihre ersten Antworten. Jetzt können wir mit der Diskussion beginnen. Jeder aus dem Publikum ist natürlich willkommen, Fragen oder Kommentare zu schreiben, und ich werde versuchen, zumindest einige davon zu lesen. Ich übergebe das Wort an Mark Taylor, der sich gemeldet hat.
Marc Taylor, ISRCTN [00:59:21]
Ich habe mich nur zu Wort gemeldet, um diese beiden Fragen zu beantworten.
Zunächst einmal zur Frage, was wir unter Veröffentlichung verstehen. Es gibt eine Richtlinie unter den Studienregistern, die von der Weltgesundheitsorganisation stammt. Und ich war an einigen Aktivitäten beteiligt, um genau zu klären, was für die Bekanntgabe der Ergebnisse ein Jahr nach Abschluss der Studie erforderlich ist, so dass die Weltgesundheitsorganisation bis zum Sommer weitere Einzelheiten dazu bekannt geben wird. Und das ist besonders wichtig, wenn es keine allgemeine Veröffentlichung dieser Ergebnisse gibt. Das ist der erste Punkt.
Punkt zwei: Haben wir eine Verringerung der Zahl der Studien festgestellt? Auf jeden Fall. Die Pandemie hatte enorme Auswirkungen auf die Forschung im Vereinigten Königreich, zum Teil, weil die Anbieter von Einrichtungen im NHS Prioritäten setzen mussten, um den Druck auf unsere Gesundheitsdienste zu mindern. Es ist also ein großes Problem, wie viele Studien durchgeführt werden und ob sie produktiv sind. Ich glaube nicht, dass der Aufwand für die Registrierung einer Studie und die Bekanntgabe der Ergebnisse das Haupthindernis für die Durchführung von Studien und die Erzielung nützlicher Ergebnisse ist.
Nano Yamasaki, HRA [01:01:24]
Was die Veröffentlichung betrifft, so möchte ich auf diese beiden Fragen zurückkommen. In dem Abschlussbericht, den wir von den Teilnehmern verlangen, stellen wir eine Reihe von Fragen. Eine der Fragen lautet: Wurde das Register aktualisiert und enthält es eine Zusammenfassung der Ergebnisse? Das ist also ein sehr spezifischer Aspekt der Oberflächengestaltung. Außerdem fragen wir, ob sich der Antragsteller an den Verbreitungsplan gehalten hat, der uns als Teil des Antrags vorgelegt wurde, und das könnte die Veröffentlichung in einer akademischen Zeitschrift oder einer Art Open-Access- Zeitschrift betreffen.
Wir legen also nicht unbedingt genau fest, welche Art von Veröffentlichung Sie vorgenommen haben. Es geht nur darum, die Fragen zu klären, die wir den Forschern und Sponsoren stellen.
Was die Auswirkungen dieser Transparenzanforderung auf die Zahl der im Vereinigten Königreich durchgeführten Studien betrifft, so würde ich mich Marks Meinung anschließen, dass es sehr schwierig ist, hier wirklich eine Ursache und Wirkung festzustellen. Ich würde sagen, dass, wenn wir einen Anstieg der Zahl der im Vereinigten Königreich durchgeführten Prüfungen feststellen, dies auf weitaus umfassendere Faktoren zurückzuführen ist und nicht unbedingt mit den Transparenzanforderungen zusammenhängt, die wir an Sponsoren und Forscher stellen. Das wäre mein Eindruck. Aber natürlich gibt es keine eindeutigen Beweise, denn wir haben keine Umfrage durchgeführt, um herauszufinden, ob die Belastung oder die empfundene Belastung durch die Anforderungen an die Forschungstransparenz die Menschen von der Durchführung von Studien abhält.
Valerie Labonte, Cochrane Deutschland (Moderation) [01:03:06]
Ich danke Ihnen. Ich habe hier eine Frage aus dem Publikum. Ich glaube, sie richtet sich auch an Naho Yamasaki. Können Sie, wenn möglich, weitere Einzelheiten zu den personalisierten Ergebnissen der Studien für die Teilnehmer erläutern? Welches Niveau streben Sie an?
Nano Yamasaki, HRA [01:03:22]
Wir fordern keine detaillierten Informationen, die der Gesundheitsforschungsbehörde zur Verfügung gestellt oder in einem Leitfaden festgehalten werden müssen. Aber was wir in diesem Bereich in Bezug auf die Strategie erwarten, ist, dass wir uns von der gemeinsamen Nutzung von Forschungsdaten und Gewebe weit entfernt haben, da wir wissen, dass in diesem Bereich bereits eine Menge Arbeit geleistet wird. Wir fragen zwar, ob sich die Leute an das gehalten haben, was sie in ihrem Antrag angegeben haben, aber wir geben keine strikten Vorgaben in diesem Bereich.
Matthias Perleth, HTA Verein [01:04:08]
Ich würde gerne noch mal auf die Traumasituation zurückkommen und warum wir hier sind. Und vielen Dank an Stefan [Sauerland], der das Problem bereits gelöst hat und uns einen Fahrplan oder einen Weg aufgezeigt hat, wie wir vorankommen können. Und ich denke, das ist irgendwie zwingend. Es ist allerdings bedauerlich, dass die deutsche Aufsichtsbehörde, das BfArM, heute nicht hier vertreten ist. Sie befinden sich natürlich in einer Situation, in der sie dem Gesundheitsministerium und dem politischen Einfluss sehr nahe stehen, und deshalb sind sie normalerweise sehr vorsichtig, nichts zu tun, was öffentlich ist. Aber natürlich müssten wir sie in die Reform einbeziehen, da sie die Registrierungspflicht wahrscheinlich tatsächlich leicht in die Praxis übertragen oder umsetzen und die Überwachung übernehmen könnten.
Was die Frage der Kosten angeht, die oft von Transparency International gestellt wird, warum nicht auf europäischer Ebene beginnen? Ich denke, das ist eine interessante Frage, und ich denke, es gibt hier wahrscheinlich zwei Aspekte. Der erste ist, dass es wahrscheinlich schneller ist, zunächst auf nationaler Ebene Fortschritte zu erzielen, anstatt darauf zu warten, dass etwas auf EU-Ebene geschieht. Zweitens bin ich mir nicht sicher, ob die Europäische Kommission oder die Europäische Union die Befugnis hat, auf nationaler Ebene Anforderungen für Interventionen zu stellen, die nicht auf EU-Ebene geregelt sind. Aber ich bin kein Jurist, also kann ich hier nur spekulieren.
[Publikumsfrage ausserhalb dieses Themas, hier ausgelassen]
Martin Smith, ehemaliger Sachbearbeiter im britischen Parlament [01:08:36]
Im Vereinigten Königreich ist die Art der gesetzlichen Grundlage, auf der die betreffende Organisation eingerichtet ist, wichtig... Der Ausschuss stellte fest, dass die Transparenz in der gesetzlichen Routine der HRA enthalten war, dass sie die Verantwortung hatte, dies zu fördern. Und das war der Schlüssel, um die Verzweiflung des Ausschusses auszudrücken, der sagte: Nun, wir halten das für unentschuldbar. Und der Test muss nach vorne gerichtet sein. Und das knüpft an das an, was Sie mit dem BfArM beschrieben haben, und die Frage, ob das formell in deren Verantwortung liegt oder nicht und ob es in deren Verantwortung gelegt werden kann, scheint eine Schlüsselfrage zu sein.
Valerie Labonte, Cochrane Deutschland (Moderation) [01:10:10]
Ich danke Ihnen vielmals. Ich habe eine Frage an die deutschen Diskussionsteilnehmer. Was denken Sie über den aktuellen politischen Willen in Deutschland? Ist der politische Wille im Moment stark genug, um mit der Arbeit an einem System zur Transparenz klinischer Studien zu beginnen?
Christoph Stein, Vorstandsmitglied von Transparency International Deutschland [01:10:35]
Ja. Nochmals, ich spreche als aktiver Forscher. Meiner Meinung nach ist der politische Wille vorhanden, und wir haben gerade alle Möglichkeiten gehört, die sehr deutlich dargelegt wurden. Ich denke, dass Deutschland bereit wäre, viele der Regeln, die im Vereinigten Königreich eingeführt wurden, umzusetzen.
Nochmals, meine Frage ist, warum sitzt die EU nicht hier? Und meine persönliche Erfahrung ist, dass wir viel mit der EU zu tun hatten. Wir mussten uns registrieren lassen. Es gibt eine große Plattform bei der EU, die ziemlich streng ist und meiner Erfahrung nach ziemlich gut funktioniert. Ich glaube also nicht, dass Deutschland allein alle anderen EU-Länder dazu drängen kann, dasselbe zu tun, so wie das Vereinigte Königreich nicht in der Lage war, dies zu tun.
Aber ich denke, wir sollten auf jeden Fall versuchen, eine gemeinsame Anstrengung zu unternehmen und einige der Instrumente zu nutzen, die bereits auf EU-Ebene existieren, und das gesamte System in der EU einzuführen.
Daniel Strech, QUEST [01:12:13]
Zum Punkt des politischen Willens. Ich meine, es ist gut von jemandem zu hören, der die Politik in dieser Hinsicht kennt, dass es dIch würde auch zustimmen, wenn die Zeit nicht jetzt ist, wann dann? Wir haben einen Gesundheitsminister, der aus der medizinischen Forschung kommt. Er ist Mediziner. Er hat einige Erfahrung in der evidenzbasierten Medizin. Und was am wichtigsten ist, er hat jetzt, nachdem Corona fast vorbei ist, etwas Zeit, um sich hoffentlich anderen Aufgaben zu widmen.
Wahrscheinlich ist es erforderlich, dass einige Interessengruppen einen gemeinsamen Brief an das Gesundheitsministerium schreiben und um eine offizielle Erklärung an das BfArM bitten, dass sie diese Aufgabe übernehmen.
Was die Frage betrifft, ob es besser ist, Universitätszentren einzubinden und die Öffentlichkeit einzubeziehen, so glaube ich nicht, dass dies wirklich gut funktioniert. Wenn man etwas rechtlich Verbindliches hat, das von einer maßgeblichen Stelle in Deutschland kommt, funktioniert das immer besser, muss ich sagen.
Stefan Sauerland, DnEBM [01:14:26]
Ich würde auch zustimmen, wenn die Zeit nicht jetzt ist, wann dann? Wir haben einen Gesundheitsminister, der aus der medizinischen Forschung kommt. Er ist Mediziner. Er hat einige Erfahrung in der evidenzbasierten Medizin. Und was am wichtigsten ist, er hat jetzt, nachdem Corona fast vorbei ist, etwas Zeit, um sich hoffentlich anderen Aufgaben zu widmen.
Wahrscheinlich ist es erforderlich, dass einige Interessengruppen einen gemeinsamen Brief an das Gesundheitsministerium schreiben und um eine offizielle Erklärung an das BfArM bitten, dass sie diese Aufgabe übernehmen.
Was die Frage betrifft, ob es besser ist, Universitätszentren einzubinden und die Öffentlichkeit einzubeziehen, so glaube ich nicht, dass dies wirklich gut funktioniert. Wenn man etwas rechtlich Verbindliches hat, das von einer maßgeblichen Stelle in Deutschland kommt, funktioniert das immer besser, muss ich sagen.
Martin Smith, ehemaliger Sachbearbeiter im britischen Parliament
Es wird Sie also nicht überraschen, dass ich dem Punkt der politischen Willensbildung voll und ganz zustimme. Ich denke, man kann sich auf das Beispiel des Vereinigten Königreichs berufen, wo sehr, sehr große Fortschritte erzielt werden können, was den Politikern die Hoffnung gibt, dass sie möglicherweise innerhalb ihres eigenen politischen Zyklus etwas bewirken können und nicht etwas, das erst in zehn Jahren zum Tragen kommen würde.
Ich würde viele der technischen Fragen beiseite lassen, die natürlich im Vordergrund stehen, z. B. wie definiert man eine Veröffentlichung und was ist mit verschiedenen Arten von Studien, die auf unterschiedliche Weise genehmigt werden? Halten Sie sich einfach an die grundlegenden Punkte über die Verschwendung von Forschungsergebnissen und die Genauigkeit der Forschungsgrundlage. Man ist den Studienteilnehmern gegenüber verpflichtet, ihre Zeit und Energie zu nutzen, indem man die Ergebnisse veröffentlicht, und dafür zu sorgen, dass das, was man untersucht, auf korrekte Art und Weise weitergeführt werden kann. Versuchen Sie einfach, sich von den Ausreden zu befreien, warum Sie in diesem Bereich nicht vorankommen, und fordern Sie, dass es eine klare Verantwortung für Fortschritte in diesem Bereich gibt.
Und wenn es dann um die technischen Details geht, können die technischen Experten in Zusammenarbeit mit der Gemeinschaft diese ausarbeiten. Aus britischer Sicht konnten wir die Tatsache nutzen, dass das britische Parlament ein starkes parlamentarisches Kontrollverfahren hat, das sehr öffentlichkeitswirksam sein kann. Der Vorsitzende des Ausschusses für Wissenschaft und Technologie war vor ein paar Jahren ein ehemaliger Minister der Regierung, ein ehemaliger Gesundheitsminister, und so hatte er sein eigenes Profil, und er wusste, was man erreichen kann, wenn man die Kontrolle nutzt, um ein Problem, das gelöst werden muss, ins Rampenlicht zu rücken. Und was auch immer das deutsche Äquivalent dazu ist, sagen Sie, dass es nicht erlaubt sein sollte, so weiterzumachen, auch wenn es technische Dinge zu klären gibt. Ich bringe den Ball jetzt ins Rollen. Das scheint mir ein sehr kluger nächster Schritt zu sein.
Naho Yamasaki, HRA [01:18:21]
Ich denke, wie Martin sagte, dass der politische Wille wirklich wichtig ist, wie auch andere kommentiert haben. Und ein gewisser Rahmen für die Durchsetzung, sei es auf rechtlichem Wege oder durch eine Agentur, die Sie haben, würde definitiv helfen.
Ich möchte aber auch betonen, dass ein mehrgleisiger Ansatz notwendig ist. Und ich denke, dass es wichtig ist, die Interessengruppen zusammenzubringen, von denen viele diese Agenda bereits unterstützt haben und den Wandel vorantreiben, und auch die Stimme der Patienten einzubringen. Ich denke, das war wirklich entscheidend, als wir die Strategie konsultiert haben. Wir haben mit vielen Bürgern gesprochen, und ich glaube, viele waren schockiert, als sie herausfanden, dass diese Art von Informationen nicht zur Verfügung steht oder nicht weitergegeben wird, dass die Ergebnisse nicht routinemäßig mit den Teilnehmern geteilt werden. Und ich denke, wir müssen diesen Zustand wirklich überwinden.
Matthias Perleth, HTA Verein [01:19:31]
Ich stimme voll und ganz mit dem überein, was gerade gesagt wurde. Ich denke, wir sollten z. B. die wichtigen Patientenvertreter einbeziehen. Einige Kampagnen in der Vergangenheit haben gezeigt, dass Patientenvertreter in Deutschland eine sehr mächtige Stimme sein können und die öffentliche Meinung beeinflussen und auch Politiker zum Handeln bewegen können, was ich vor ein paar Tagen im Fall von Long Covid in Deutschland gesehen habe. Und ich denke, dass dies einige Maßnahmen auslösen kann.
Ich denke, es würde wahrscheinlich nicht ausreichen, nur einen Brief an einige Entscheidungsträger zu schreiben. Aber ich würde die Briefaktion als einen Teil einer koordinierteren Kampagne betrachten, die versucht, eine andere Strategie anzuwenden, zu der natürlich auch Briefe an Entscheidungsträger gehören. Was wir jedoch brauchen, ist ein politischer Meinungsführer, idealerweise im Ministerium, vielleicht der Minister selbst oder eine andere hochrangige Persönlichkeit oder ein Mitglied des Parlaments. Und in einem der Ausschüsse des Deutschen Bundestages müssen wir sie, so würde ich sagen, in Brand setzen, um für dieses Thema zu werben.
Und wir sollten die Vorteile der Einführung einer Registrierungspflicht betonen. Und wir sollten wahrscheinlich auch betonen, dass es nichts Schlechtes daran gibt. Es gibt also keine finanziellen Probleme. Niemand kann hier etwas verlieren. Und ich denke, es ist wichtig, dies im Auge zu behalten. Es geht nicht darum, Geld zu verteilen oder ähnliches. Es geht nur um Transparenz. Und daran ist nichts Schlechtes.
Valerie Labonte, Cochrane Deutschland (Moderatorin) [01:21:52]
Ich würde gerne eine Frage aus dem Publikum vorlesen. Ich habe nie verstanden, warum Ethikkommissionen die Registrierung und Veröffentlichung nicht überprüfen. Ist es ein Mangel an Ressourcen? Ist das die einzige Einschränkung? Möchte jemand auf diese Frage antworten?
Christoph Stein, Vorstandsmitglied von Transparency International Deutschland [01:22:11]
Auch hier kann ich aus eigener Erfahrung sagen, dass sich die Ethikkommissionen in der Regel nicht mit dieser Frage befassen. Aus irgendeinem Grund konzentrieren sie sich ausschließlich auf den Beginn der Studie, auf die Belastung für die Patienten. Und mir persönlich wurde noch nie die Frage nach der Veröffentlichung gestellt, und ich weiß nicht, warum das so ist, aber ich denke, das sollte so
sein. Dem stimme ich voll und ganz zu. Es sollte zu ihren Pflichten gehören, über die Veröffentlichung nachzudenken, keine Frage.
Vielleicht müssen die Regeln, die Gesetze für Ethikkommissionen geändert werden. Das wäre eine Möglichkeit. In Deutschland ist das nicht so einfach, weil wir sehr viele verschiedene Ethikkommissionen haben. Wir haben Ethikkommissionen auf Landesebene. Wir haben in Deutschland 16 Bundesländer. Jedes Bundesland hat also seine eigene Ethikkommission, und wir haben Ethikkommissionen auf institutioneller Ebene. Wenn Sie also nur die Universitätskliniken nehmen, haben wir in Deutschland etwa 36 Universitätskliniken. Zählt man diese zu den Landes- Ethikkommissionen hinzu, kommt man auf etwa 50 verschiedene Ethikkommissionen, und da sind die lokalen Ethikkommissionen der kommunalen Krankenhäuser noch gar nicht eingerechnet.
In Deutschland ist es also immer sehr schwierig, ein allgemeines Gesetz zu schaffen, das für alle verschiedenen Einrichtungen und Bundesländer verbindlich ist. Aber es sollte auf jeden Fall ein Gesetz sein.
Daniel Strech, QUEST [01:24:10]
Wir würden es gerne sehen, wenn alle interventionellen Studien gemäß der Deklaration von Helsinki registriert würden, aber das ist nicht wirklich Teil des deutschen Gesetzes, Stefan. Es ist Teil der Position des Arztes, weiches Recht. Was also passieren könnte, ist, dass ein Arzt seine Approbation verliert, aber das ist nicht wirklich das Stadium, in dem wir uns befinden.
Es ist jetzt zehn Jahre her, dass ich eine Gruppe für diesen Dachverband der deutschen Forschungsethikkommissionen [AKEK] geleitet habe. Was würden die etwa 50 Forschungsethikkommissionen in Deutschland über die Registrierung von Studien für alle Studien denken, nicht nur für Arzneimittelstudien und vielleicht in Zukunft auch für Studien mit Medizinprodukten? Und Sie finden diese [AKEK]-Stellungnahme, dieses Meinungsbild aus dem Jahr 2012 immer noch auf deren Website, das dies im Prinzip unterstützt. Das Hauptargument am Ende der Stellungnahme ist, dass wir als Ethik-Kommissionen in Deutschland keine rechtliche Grundlage dafür haben. Wir können das nicht einfach verlangen. Das gilt nur für die Arzneimittelprüfungen.
Wenn es um die Berichterstattung über die Ergebnisse geht, ist es mehr oder weniger die gleiche Geschichte. Es ist sogar noch etwas komplizierter, denn die Registrierung von Studien könnte im Prinzip bei der Genehmigung verlangt werden, wie wir es bei allen Arzneimittelprüfungen tun. Aber um die Berichterstattung über die Ergebnisse im Rahmen des Ethikantrags zu berücksichtigen, müssten Sie zumindest eine Rückmeldung darüber erhalten, ob die abgeschlossenen Studien etwas veröffentlicht haben oder nicht, zum Beispiel 12 Monate nach dem Abschlussdatum, und dann auf sie zurückkommen. Und dann kann man die Genehmigung der Studie nicht verweigern, aber man kann vielleicht versuchen, bestimmte Anreize zu geben, damit die Ergebnisse trotzdem veröffentlicht werden usw. Die Ethikkommission hat also derzeit einen rechtlichen Hintergrund, abgesehen von der Tatsache, dass es ihr derzeit zumindest an Ressourcen, an persönlichen finanziellen Mitteln fehlt, um ein solches Verfahren zu entwickeln.
Aber wie die britische Initiative zeigt, kann man das wirklich zentralisieren, man kann Dinge automatisieren und mit ein oder zwei rechtlichen Unterstützungsmechanismen kann man dann diese Infrastruktur aufbauen. Wir werden alles automatisch registrieren lassen, und man kann zumindest automatisch generierte Erinnerungsschreiben 12 Monate nach Abschluss der Studie verschicken. All dies sollte im Prinzip keine Raketenwissenschaft sein. Es wäre prinzipiell möglich. Aber die Ethikkommissionen allein können das nicht umsetzen. Sie müssen mit einigen Leuten zusammenarbeiten, die den politischen Willen und die politische Entscheidungsgewalt haben.
Valerie Labonte, Cochrane Deutschland (Moderatorin) [01:27:13]
Ich sehe, die Zeit läuft uns davon. Ich danke Ihnen allen für eine sehr lebhafte Diskussion. Ich hoffe, dass dieses Treffen die richtigen Leute zusammengebracht hat, um den Prozess zur Schaffung eines Studientransparenzsystems für Deutschland einzuleiten. Jetzt bin ich gespannt, wie es weitergeht. Ich danke Ihnen allen, dass Sie sich heute Zeit genommen haben. Es war mir ein echtes Vergnügen. Ich danke Ihnen.
[ENDE]
Dieses Transkript wird unter einer Creative Commons Lizenz (CC-BY 4.0) veröffentlicht.