
RESEARCH
Human and financial consequences of herding in oncology drug development: clinical trials of TIGIT inhibitors
(BMJ Oncology, 2026)
Emily Zhang, Leeza Osipenko, John Hickman
In oncology drug development, herding—where five or more biopharmaceutical companies compete to develop modulators of the same target—dramatically increases the number of patients exposed to experimental therapies and inflates trial costs, as illustrated by the case of TIGIT (T-cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains) inhibitors. Analysis of data from public registries like ClinicalTrials.gov and the International Clinical Trials Registry Platform revealed that 21 companies pursued approval for 30 TIGIT inhibitors, resulting in 220 clinical trials that enrolled nearly 49,000 patients between May 2016 and September 2025, at an estimated total cost to companies of US$3.1–3.6 billion. By November 2025, development of 8 of these 30 inhibitors (27%) had been terminated due to lack of significant clinical activity or strategic decisions, involving nearly 15,000 patients in those failed efforts, and none of the TIGIT inhibitors had received marketing authorization. Given the high attrition rate (~95%) typical in oncology, this herding phenomenon amplifies substantial human and financial losses by concentrating resources on a narrow set of similar candidates, diverting investment from a more diversified portfolio that could better spread risk and potentially yield more successful therapies.


Barriers to and facilitators of clinical trial participation in multiple myeloma
(Blood Global Hematology 2026)
Martin Kaiser, Saba Ul-Hasan, Jack Lewis, Federica Mirto, Alicia O’Neill, Yelak Biru, Cynthia Chmielewski, Katie Joyner, Elise Gamertsfelder
Multiple myeloma (MM) remains an incurable malignancy despite significant therapeutic advances, and disparities in healthcare outcomes persist globally. While clinical trials are the foundation for new interventions, recruitment barriers often result in unrepresentative patient groups, depriving many of access to experimental treatments. To investigate these challenges, geographic data from MM trials registered between 2011 and 2021 were examined, alongside interviews with international stakeholders, including patients and healthcare professionals.
Analysis of the data highlighted significant global inequality, with 61% of identified trials recruiting patients from the United States, while other regions remained underrepresented. Through stakeholder interviews, critical barriers were identified, including overly stringent eligibility criteria, a lack of trial awareness, and various logistical challenges. To address these issues, a set of globally applicable recommendations was developed. Unanimous support was found for broadening eligibility criteria to better reflect real-world populations, alongside calls for enhanced patient education, improved physician communication, and increased international collaboration to strengthen research infrastructure.
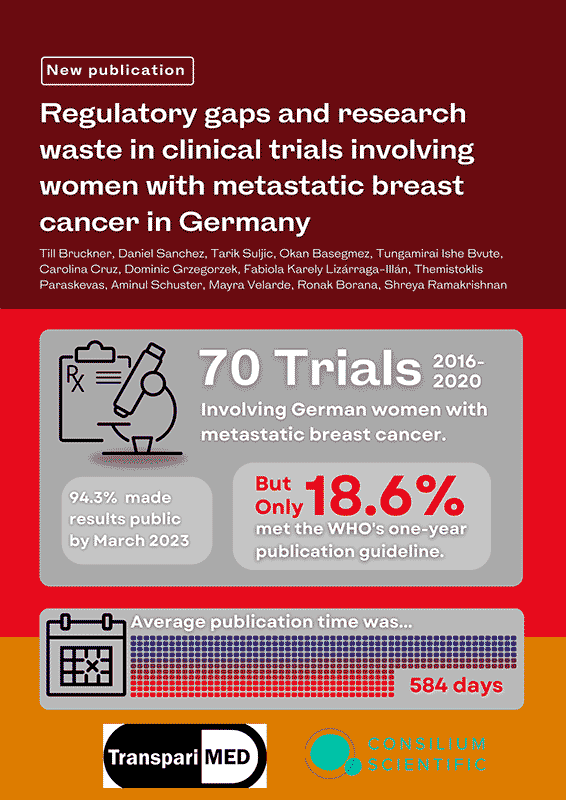
Regulatory gaps and research waste in clinical trials involving women with metastatic breast cancer in Germany
(F1000 Research, 2024)
Till Bruckner, Daniel Sanchez, Tarik Suljic, Okan Basegmez, Tungamirai Ishe Bvute, Carolina Cruz, Dominic Grzegorzek, Fabiola Karely Lizárraga-Illán, Themistoklis Paraskevas, Aminul Schuster, Mayra Velarde, Ronak Borana, Shreya Ramakrishnan
Non-publication and delayed publication of clinical trial results contribute significantly to research waste and ethical violations in medical research. The Declaration of Helsinki and the World Health Organisation (WHO) mandate timely public reporting of interventional clinical trial results to enhance the medical evidence base and prevent harm to patients. Despite these guidelines, substantial gaps in the reporting of clinical trials persist.This study focuses on clinical trials involving German women with metastatic breast cancer—a critical area given the high incidence and mortality rates associated with the disease. The research assesses both the speed and completeness of reporting results across trials sponsored by various entities and governed by different regulatory frameworks.A cohort of 70 trials completed or terminated between 2016 and 2020 was analysed. The findings reveal that only 13 of these trials (18.6%) reported results within the one-year timeframe recommended by the WHO. Furthermore, by March 2023, results for only 66 of the 70 trials (94.3%) had been made public, leaving a small but significant fraction unreported.This research not only documents the current state of clinical trial reporting in Germany but also underscores the urgent need for comprehensive legislative and regulatory reforms to ensure full transparency. The aim is to minimise research waste and improve patient outcomes by facilitating the development of more effective and safer medical interventions for metastatic breast cancer.

Assessment of quality of data submitted for NICE technology appraisals over two decades
(BMJ Open, 2024)
by Leeza Osipenko, Saba Ul-Hasan, Debra Winberg, Kseniia Prudyus, Marina Kousta, Artemis Rizoglou, Isabella Rustignoli, Laurens van der Maas
The National Institute for Health and Care Excellence (NICE) pioneered the Health Technology Assessment (HTA) processes and methodologies. Technology appraisals (TAs) focus on pharmaceutical products and clinical and economic data, which are presented by the product manufacturers to the NICE appraisal committee for decision-making. Uncertainty in data reduces the chance of a positive outcome from the HTA process or requires a higher discount.
In this work we investigate the quality of clinical data (comparator, quality of life (QoL), randomised controlled trials (RCTs) and overall quality of evidence) submitted by the manufacturers to NICE.
409 TAs were analysed (multiple technology appraisals (MTA)=104, single technology appraisal (STA)=305). In two-thirds of TAs, the overall quality of evidence was either poor (n=224, 55%) or unacceptable (n=41, 10%). In 39% (n=119) of the STAs, the quality
of comparative evidence was considered poor, and in 17% (n=51) unacceptable. In 44% (n=135) of STAs, the quality of QoL data was considered poor, 15% (n=47) unacceptable, 33% (n=102) acceptable and 7% (n=21) as good. Over 20 years of longitudinal analysis did not show improvements in the quality of evidence submitted to NICE.
We found that the primary components of clinical evidence influencing NICE’s decision-making framework were of poor quality. It is essential to continue to generate robust clinical data for premarket and post-market introduction of medicines into clinical practice to ensure they deliver benefits to patients.
In the press:
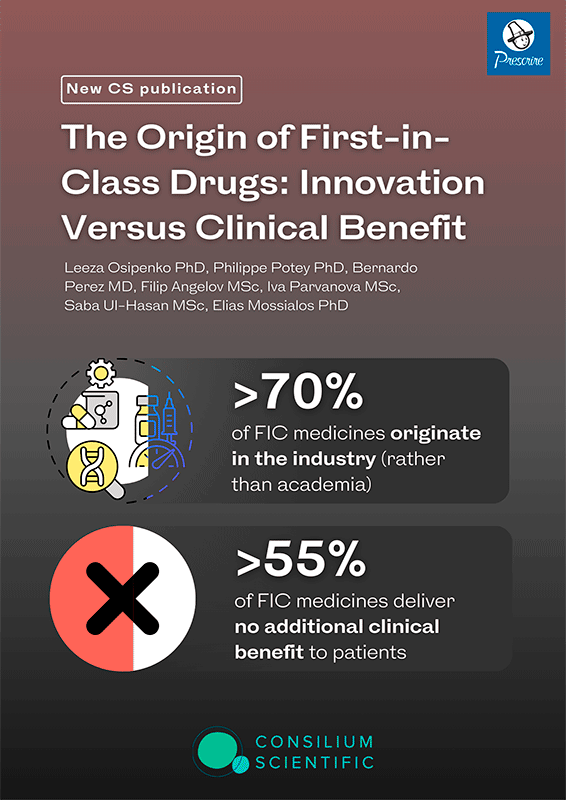
The Origin of First-in-Class Drugs: Innovation Versus Clinical Benefit
(Clin Pharmacology & Therapeutics, 2023)
by Leeza Osipenko, Philippe Potey, Bernardo Perez, Filip Angelov, Iva Parvanova, Saba Ul-Hasan, Elias Mossialos
First-in-class (FIC) designation became a hallmark of innovation, however, even at the marketing authorization stage, little is known about the clinical benefits these products deliver. We identified the provenance of the FIC drugs that entered the French market from 2008 to 2018 and matched these medicines to the clinical benefit grading by Haute Autorité de Santé (HAS) and Prescrire. Analyses were performed using descriptive statistics to present our findings by drug origin and therapeutic area and to establish the degree of concordance between HAS and Prescrire. Of the 135 FIC drugs identified, 71.1% (n = 96) originated from the industry, 16.3% (n = 22) from academia, and 12.6% (n = 17) from joint partnerships. Three therapeutic areas accounted for most FIC medications: antineoplastic (25.9%, N = 35), anti-infective (14.1%, N = 19), and metabolic (11.1%, N = 15) agents. HAS and Prescrire agreed on 60.74% of clinical benefit gradings. According to HAS, only 5% of all FIC drugs had substantial added benefit, and only 3%, according to Prescrire. HAS and Prescrire graded 45.9% and 68.2%, respectively, of FIC drugs as no clinical benefit and 48.9% and 28.9%, respectively, as some clinical benefit. FIC-designated drugs are primarily of industry (> 70%) rather than academic origin. We found that 55% of FIC medicines that entered the French market over the 10-year period deliver no additional clinical benefit. Whereas FIC medicines may represent important scientific advancements in drug development, in > 50% of cases, the new mode of action does not translate into additional clinical benefits for patients.
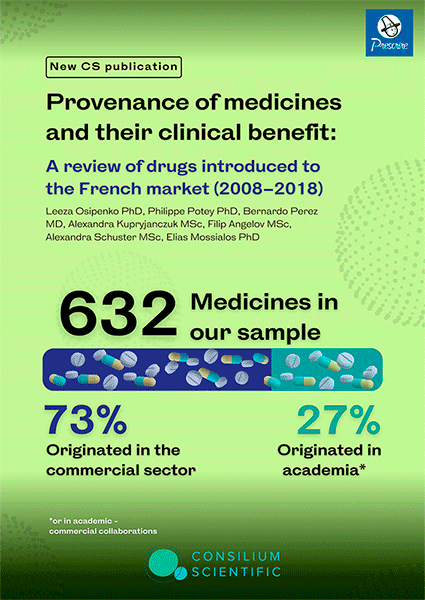
Provenance of medicines and their clinical benefit: A review of drugs introduced to the French market (2008–2018)
(JAMA Internal Medicine, 2023)
by Leeza Osipenko, Philippe Potey, Bernardo Perez, Alexandra Kupryjanczuk, Filip Angelov, Alexandra Schuster, Elias Mossialos
Both the commercial sector and academia play a vital role in medicine development. Ongoing debates exist on their contribution and the value of medicinal products entering the market. In this work we identify the provenance and clinical benefit of medicines that entered the French market between 2008 and 2018. Of the 632 medicines that entered the French market between 2008 and 2018, 73% originated in the commercial sector, and 27% originated in the academic setting or in collaboration with commercial enterprises. Prescrire graded psychotropic agents (93%; 13/14), while HAS graded respiratory agents (96%; 24/25) as the highest percentage of medicines that provided no added benefit. Prescrire graded 77.6% of medicines that originated in the industry and 64.3% that originated in the academic setting (p=0.001) to have no added clinical benefit. HAS assigned such grading to 71.3% (industry) vs 61.9% (academia) (p=0.024). Based on the Prescrire grading, academia invented more medicines delivering some added benefit 33.9% vs 21.12% invented by industry (p=0.001). HAS grading on some added benefit 30.4% (academia) vs 26.1% (industry) did not reach statistical significance (p=0.29). However, HAS grading on substantial added clinical benefit reached statistical significance in favour of academia 7.7% vs 2.6% in the industry (p=0.003), while Prescrire grading did not (1.8% academia vs 1.3% industry p=0.64). Over 70% of medicines that entered the French market during the 10-year period originated in the commercial sector. Although the majority of medicines were not graded as providing clinical benefit, medicines originating in the academic setting were more likely to be graded as conferring clinical benefit than those originating in the commercial setting.
Evaluating Clinical Trials Transparency Policies at UK Public Research Institutions
by Dr. Nicholas J. DeVito and Scott Lewis
In this work we establish that:
1. Transparency Requirements should be concrete, universal, and come directly from the institution. Transparency policies should have strong, unambiguous language that establishes internal institutional requirements.
2. The processes for transparency practice should be described in detail. Policies should contain a clear articulation of who is responsible for these activities, what exactly those responsibilities entail, and how details of how to carry them out.
3. The monitoring or auditing of transparency practices should be documented with specific and well-described escalation pathways, responsible parties, and potential penalties for non-compliance.
Ongoing monitoring is key to ensuring transparency responsibilities are being met. Sufficient documentation on how institutions plan to implement these activities will offer clarity and set expectations for researchers and staff.


Costs and Causes of Oncology Drug Attrition With the Example of Insulin-Like Growth Factor-1 Receptor Inhibitors
(JAMA Network Open, 2023)
By Valerie Jentzsch, Leeza Osipenko, Jack Scannell, John Hickman
In this paper we are aiming to answer the following question: what are the associated expenses of clinical research and what factors underly the translational failure of inhibitors of the insulin-like growth factor-1 receptor (IGF-1R) in oncology? Our findings show that 16 inhibitors of IGF-1R underwent 183 clinical trials in more than 12 000 patients; none of the agents was approved for clinical use in oncology practice and the trials were estimated to have had expenses of greater than $1.6 billion. Half of the published in vivo preclinical data analyzed showed less than a 50% inhibition of tumor growth by IGF-1R inhibitors. With high attrition rates for oncology drugs, the fruitless and expensive clinical trials of 16 IGF-1R inhibitors draw attention to the need for improved preclinical models and better decision-making before trials are launched, reducing substantial financial losses and avoiding exposure of patients to potential toxic effects.
Read full text | Watch video | Podcast
Invited Commentary
Journeys to Failure that Litter the Path to Developing New Cancer Therapeutics
Tito Fojo, MD, PhD
Endpoints News: Failed trials for 16 IGF-1R inhibitors and $1.6B later: Time for better preclinical models? by Zachary Brennan
Cancer Therapy Advisor: Failed Oncology Trials May Cost Up to $60 Billion Per Year by Hibah Khaja
Towards transparency: adoption of WHO best practices in clinical trial registration and reporting among top medical research funders in the USA
(BMJ EBM, 2023)
Elise Gamertsfelder, Netzahualpilli Delgado Figueroa, Sarai Keestra, Alan Rossi Silva, Ronak Borana, Maximilian Siebert, Till Bruckner
In this work we assess to what extent the clinical trial policies of the largest public and philanthropic funders of clinical research in the United States meet WHO best practices in trial registration and reporting.
Fourteen funders were assessed. Our cross-sectional study found that, on average, funders have only implemented 4.1/11 (37%) of WHO best practices in clinical trial transparency. The most frequently adopted requirement was open access publishing (14/14 funders). The least frequently adopted were (1) requiring trial ID to appear in all publications (2/14 funders, 14%) and (2) making compliance reports public (2/14 funders, 14%). Public funders, on average, adopted more policy elements (5.2/11 items, 47%) than philanthropic funders (2.8/11 items, 25%). Only one funder’s policy documents mentioned the CONSORT statement.
There is a significant variation between the number of best practice policy items adopted by medical research funders in the USA. Many funders fell significantly short of WHO Joint Statement benchmarks. Each funder could benefit from policy revision and strengthening.
Are European clinical trial funders policies on clinical trial registration and reporting improving? A cross-sectional study
(J Clin Transl Sci, 2023)
by Marguerite O’Riordan, Martin Haslberger, Carolina Cruz, Tarik Suljic, Martin Ringsten, Till Bruckner
In this study we assess the extent to which the clinical trial registration and reporting policies of 25 of the world’s largest public and philanthropic medical research funders meet best practice benchmarks as stipulated by the 2017 WHO Joint Statement, and document changes in the policies and monitoring systems of 19 European funders over the past year.
This study enables funders worldwide to identify and address gaps in their clinical trial transparency policies by pinpointing exactly where they currently fall short of WHO best practices. It also enables policymakers and citizens to assess whether public bodies tasked with furthering medical knowledge have adopted adequate safeguards against research waste and publication bias.
Data redaction practices in NICE technology appraisals from 1999 to 2019
(BMJ Open, 2021)
This paper presents a complete audit of data redaction practices in all active technology appraisals (TA) and highly specialised technology (HST) evaluations issued by the National Institute for Health and Care Excellence (NICE) from the conception of the Institute to September 2019. Over 81.6% of appraisals feature data redactions. Data redaction increased substantially over time, and is currently at its highest level with 100% of TAs having at least some data redaction in 2019/2020, 96% of appraisals in 2018/2019% and 94% of appraisals in 2017/2018. Policy changes are needed to stop clinical data redaction practice at NICE.
UK government refuses to discuss ‘alarming’ NICE data redactions
Pressure Builds On England’s HTA Body & Industry To Reduce Data Redaction
ICMJE statement on clinical trial data contained in assessment reports
Health groups call for end to redacted clinical trial data in technology assessments
BMJ News Jan 19, 2022
Feb 16, 2022
Why NICE’s Lack of Data Transparency Undermines Priority Setting in Low- and Middle- Income Countries
The European Union and personalised cancer medicine
(EJC, 2021)
By John Hickman, Ian Tannock, Lisa Hutchinson and Lydie Meheus
This paper discusses two recent policy documents by the European Union, ‘Europe’s Beating Cancer Plan’ and its accompanying ‘Conquering Cancer: Mission Possible’. These articulate broad policies aimed at reducing cancer mortality across Europe. The focus for cancer treatment in these manifestos is the expansion of personalised cancer medicine (PCM). We address the limits of PCM in pathology-driven and pathology-agnostic PCM, briefly discussing the results of umbrella and basket trials. We suggest that the complexity, plasticity and genetic heterogeneity of advanced cancers will continue to thwart the impact of PCM, limiting it to specific pathologies, or rare subsets of them, and that caution regarding the advancement of PCM is justified. Policymakers should be wary of the hype of lobbyists, who do not acknowledge the limits of PCM.
Blockchain’s potential to improve clinical trials
(BMJ, 2019)
“Blockchain” has become a buzzword. Some entrepreneurs and researchers evangelise its promise of innovation, but others are turned off by the hype surrounding the technology. Blockchain sceptics warn about high costs. There’s more to this tamperproof technology than bitcoin. It could be used to improve the administration of clinical trials, ensuring transparency and yielding better quality data. Patients taking part in clinical trials deserve every record to be counted, appropriately handled, and independently assessed. Increased transparency and data integrity would help to accomplish this. The remit of regulators and the funders of non-commercial research is to protect and advance public health—and therefore they, rather than industry or academia, should lead blockchain’s implementation in managing clinical trials.
Listen to podcast here
Limits to Personalized Cancer Medicine
(NEJM, 2016)
By John Hickman and Ian Tannock
This commentary addresses some of the reasons for the limited impact of cancer drugs targeted to the genetic lesions driving malignancies. The genetic heterogeneity of advanced cancers is a particular barrier to the effective use of single targeted therapies because there are multiple genetic drivers. The paper reviews some of the data on the use of drug combinations that attempt to overcome the challenge of genetic heterogeneity, suggesting that drug combinations are limited by significant host toxicity. In particular, the paper questions the growing use of targeted drugs guided by the next generation sequencing of tumour genomes, in so-called precision or personalised medicine trials. It asks that this approach is only taken as part of well-controlled trials and that patients should be informed of the limits of personalised cancer medicine with targeted therapies.